- РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ веществ
- ОФС.1.2.1.2.0001.15 Хроматография
- ОФС.1.2.1.2.0001.15 Хроматография
- ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
- интерпретация хроматографических данных
- РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
- Метод нормирования (метод внутренней нормализации).
- Метод внешнего стандарта.
- Метод внутреннего стандарта.
- Метод стандартных добавок.
- РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
- ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
- Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
- КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
- Тонкослойная и бумажная хроматография
- Жидкостная хроматография: изократическое элюирование
- Жидкостная хроматография: градиентное элюирование
- Газовая хроматография
- Сверхкритическая флюидная хроматография
- Хроматографические методы анализа. Учебно-методическое пособие (стр. 3 )
- Характеристики удерживания
- 🎬 Видео
Видео:Расчет RRF и анализ образцаСкачать

РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ веществ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют четыре основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
1. Метод нормирования (метод внутренней нормализации).
Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:
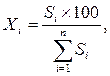
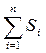 – сумма площадей (высот) всех пиков на хроматограмме.
– сумма площадей (высот) всех пиков на хроматограмме.
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.
Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле
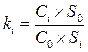 ,
,
где: Сi и С0 – концентрация i-ого вещества и стандартного вещества, соответственно;
Si и S0 – площадь (высота) пика i-ого вещества и стандартного вещества, соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8–1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:
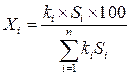 .
.
При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8–1,2.
2. Метод внешнего стандарта. Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах стандартного раствора.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:
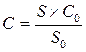 ,
,
где: S и S0– средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов, соответственно;
С и С0 – концентрации определяемого и стандартного растворов, соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве стандартного раствора для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в стандартном растворе по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8-1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой,в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
3. Метод внутреннего стандарта. Концентрация определяемого вещества определяется путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора, и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме стандартного раствора. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле
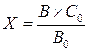 ,
,
где: 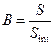 – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
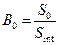 – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в стандартном растворе;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в стандартном растворе;
С0 – концентрация определяемого вещества в стандартном растворе.
В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
4. Метод стандартных добавок. Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условий с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:
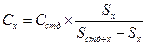
где: Сстд концентрация стандартной добавки;
Sx интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
Видео:Хроматэк Аналитик 3.1. ИнтегрированиеСкачать

ОФС.1.2.1.2.0001.15 Хроматография
Содержимое (Table of Contents)
Видео:Хроматэк Аналитик 3.1. Расчёт среднихСкачать

ОФС.1.2.1.2.0001.15 Хроматография
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Хроматография ОФС.1.2.1.2.0001.15
Взамен ст. ГФ XI, вып.1
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения. По механизму, лежащему в основе разделения, различают адсорбционную, распределительную, ионообменную и другие виды хроматографии.
В настоящее время используются следующие хроматографические методы анализа, представленные на рис.1

Рисунок 1- Методы хроматографического анализа.
Результат хроматографического разделения представляется в виде хроматограммы.
Видео:Хроматэк Кристалл: Детектор ПФДСкачать

ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Хроматограмма представляет собой графическое или иное представление сигнала детектора, концентрации веществ в элюате или другой количественной величины, используемой для измерения концентрации веществ в элюате, от времени или объема подвижной фазы. В планарной (плоскостной) хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или ТСХ-пластинке (тонкослойная хроматография) последовательность зон адсорбции веществ исходной (анализируемой) смеси.
Схематически хроматограммы представляют собой последовательность гауссовых пиков на базовой линии (рис. 2).
Базовая линия – сигнал от подвижной фазы.
Пик – часть хроматограммы, регистрирующая отклик детектора. Пик отображает постепенное нарастание концентрации вещества и последующее ее уменьшение. В случае линейной изотермы сорбции кривая, описывающая пик, приближается к кривой гауссова распределения.
Основание пика — продолжение базовой линии, соединяющее начало и конец пика.
Площадь пика (S) – площадь хроматограммы, заключенная между кривой, описывающей пик, и его основанием.
Высота пика (H) – расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора.

Рисунок 2 — Хроматограмма и основные хроматографические параметры: 1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося вещества; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)
Видео:Хроматэк Аналитик 3.1. Создание метода с нормализациейСкачать

интерпретация хроматографических данных
Время удерживания (tR или t) – время, необходимое для элюирования вещества. Соответствует времени появления максимума пика на хроматограмме.
Объем удерживания (VR) – объем подвижной фазы, необходимый для элюирования вещества. Может быть вычислен по времени удерживания и скорости потока (F).

Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока.
Время удерживания несорбирующегося вещества (t0 или tм) – время, необходимое для элюирования неудерживаемого на сорбенте вещества. В эксклюзионной хроматографии t0 соответствует времени удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Объем удерживания несорбирующегося вещества (V0) – объем подвижной фазы, необходимый для элюирования неудерживаемого вещества. Может быть вычислен по времени удерживания неудерживаемого вещества и скорости потока (F):

В эксклюзионной хроматографии V0 соответствует объему удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Общее время удерживания подвижной фазы (tt) – в эксклюзионной хроматографии время удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Общий объем удерживания подвижной фазы (Vt) – в эксклюзионной хроматографии объем удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Константа (коэффициент) распределения (K0) – в эксклюзионной хроматографии характеристика элюирования вещества из определенной колонки, которую рассчитывают с помощью выражения:

Приведенное (исправленное) время удерживания вещества (t´R) ‒ время удерживания вещества за вычетом времени удерживания несорбируемого вещества. Может быть рассчитано по формуле:

Исправленное время удерживания не зависит от объема трубопроводов хроматографической системы, установленных между инжектором и колонкой.
Относительное время удерживания (r) – относительное приведенное (исправленное) время удерживания вещества 2 по веществу 1:

Нескорректированное относительное время удерживания (RRT) – относительное время удерживания вещества 2 по веществу 1:

Если не указано иное, значения относительного времени удерживания, приведенные в фармакопейных статьях, соответствуют нескорректированному относительному времени удерживания
Коэффициент емкости (k´) – коэффициент емкости колонки по веществу с временем удерживания tR, показывающий, во сколько раз исправленное время удерживания вещества больше, чем время удерживания несорбируемого вещества.

Эффективность хроматографической системы ‒ параметр, характеризующий степень размывания хроматографического пика. Эффективность выражается числом теоретических тарелок (N):


W – ширина пика у основания;
W0,5 – ширина пика на половине высоты.
При расчете числа теоретических тарелок значения времени удерживания и ширины пика должны быть приведены к одинаковой размерности.
Число теоретических тарелок зависит от природы определяемого вещества, его концентрации или объема, вводимого в систему, от колонки, температуры колонки и состава подвижной фазы.
Если не указано иное, то эффективность хроматографической системы, требования к которой приведено в фармакопейной статье, рассчитывается по формуле, использующей ширину пика на половине его высоты.
Фактор асимметрии (фактор симметрии) пика (As) рассчитывают по формуле:

W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты (рис. 3).
Если фактор асимметрии равен 1, то пик симметричен. Если фактор асимметрии больше 1, то это означает, что растянут задний фронт пика. Если фактор асимметрии меньше 1, то пик растянут спереди.

Рисунок 3 — Схема расчета фактора асимметрии пика
Разрешение (Rs).
Разрешение между пиками двух веществ смеси элюирующимися друг за другом рассчитывают по формулам:


при этом tR2 ≥ tR1. При расчете разрешения величины времени удерживания и ширины пиков должны быть приведены к одинаковой размерности.
Если не указано иное, то разрешение, требования к которому приведены в фармакопейной статье, рассчитывается по формуле, использующей ширину пиков на половине высоты.
В случае, если пики несимметричны и если интенсивность пиков значительно различается, параметр Rs не всегда корректно описывает разделение хроматографических пиков. Таким образом, даже при значениях Rs ≥ 1,5 может наблюдаться неполное разделение пиков. В этих случаях при оценке разделяющей способности можно заменить параметр Rs на параметр «отношение максимум/минимум».
Отношение максимум/минимум (p/v), называемое также отношением «peak-to-valley», «пик – долина». Этот параметр позволяет оценить разделительную способность хроматографической системы. Значение p/v рассчитывается по формуле:

Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии (рис. 4).

Рисунок 4 — Хроматограмма не полностью разделяемых веществ
Данное соотношение применяется для оценки разделительной способности хроматографической системы, если вещества смеси разделяются не полностью. Рассчитанное отношение максимум/минимум в значительной степени зависит от выбранного варианта интегрирования хроматограммы. Результаты измерения соотношения p/v будут некорректны в случае разметки меньшего пика методом экстраполяции смещенной базовой линии (методом тангенциальной касательной).
Отношение сигнал/шум (S/N).
Краткосрочный шум сигнала детектора (шум базовой линии) влияет на прецизионность количественного определения. Отношение сигнал/шум рассчитывают по формуле:

Н – высота пика, соответствующего рассматриваемому веществу на хроматограмме указываемого стандартного раствора, измеренная от максимума пика до экстраполированной базовой линии. Экстраполяция базовой линии проводится для сигнала на участке базовой линии во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте;
h – размах фонового шума, измеряемый либо на хроматограмме контрольного (холостого) раствора (или раствора плацебо), либо на хроматограмме того же раствора стандартного образца.
Измерение размаха фонового шума проводится во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте, расположенном, если это возможно, равномерно по обе стороны от места возможного обнаружения пика. В случае использования для измерения шума хроматограммы контрольного (холостого) раствора (или раствора плацебо) измерение размаха фонового шума проводится во временном интервале, включающем в себя время удерживания рассматриваемого вещества, при этом продолжительность временного интервала, в котором проводится измерение шума, должна не менее чем в 5 раз превышать ширину на половине высоты для пика на хроматограмме указываемого раствора стандартного образца (рис. 5).

Рисунок 5 — Вычисление отношения сигнал/шум.
Объем задержки (D) (объем задержки градиента) представляет собой объем системы между точкой, в которой происходит смешение элюентов, и началом колонки.
Объем задержки градиента влияет на времена удерживания веществ и общий профиль наблюдаемой хроматографической картины, получаемой при использовании градиентного элюирования. Объем задержки хроматографической системы определяется следующим способом.
Колонка. Заменяют хроматографическую колонку капилляром (например, 1 м × 0,12 мм).
Подвижная фаза.
Подвижная фаза A — вода.
Подвижная фаза B — 0,1 % (об/об) раствор ацетона.
| Время, мин | Подвижная фаза A, % (об/об) | Подвижная фаза B, % (об/об) |
| 0 – 20 | 100 → 0 | 0 → 100 |
| 20 – 30 | 0 | 100 |
Скорость потока. Необходимая для создания давления, достаточного для стабильной работы насоса (например, 2 мл/мин).
Детектор. Спектрофотометрический при длине волны 265 нм.
Определяют время (t0,5) в минутах, когда оптическая плотность увеличилась на 50 %. Вычисляют объем задержки:
tG – предварительно установленное время градиента (20 мин);
F –скорость потока, мл/мин.

Рисунок 6 — Определение объема задержки градиента
Прецизионность системы в условиях повторяемости
Прецизионность системы в условиях повторяемости выражается в виде рассчитанного относительного стандартного отклонения в процентах [RSD (%)] по результатам последовательных измерений не менее чем 3 вколов или нанесений раствора сравнения и рассчитывается по формуле:

– среднее значение результатов определений;
– результаты единичных определений;
n – число определений.
В планарной хроматографии аналогом времени удерживания является фактор удерживания (Rf):

где a – расстояние от точки нанесения пробы до центра пятна, характеризующего зону адсорбции;
b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют условия хроматографирования. Оценкой хроматографической подвижности, менее чувствительной к влиянию отклонений в условиях проведения эксперимента, является величина Rst, представляющая собой отношение величины Rf одного вещества к величине Rf другого, принятого за стандарт:

Величины Rf и Rst используют для идентификации веществ. Обычно выбор стандарта осуществляется так, чтобы значение Rst анализируемого вещества было в пределах от 0,5 до 2,0. Схема определения этих величин приведена на рис. 7. 
Рисунок 7 — Схема определения значений Rf и Rst.
 – место нанесения образца на линию старта; 1 – анализируемое вещество (а); 2 – вещество-стандарт (ст); b – расстояние от линии старта до линии фронта элюента.
– место нанесения образца на линию старта; 1 – анализируемое вещество (а); 2 – вещество-стандарт (ст); b – расстояние от линии старта до линии фронта элюента.
Данные планарной хроматографии могут быть представлены в виде денситограмм.
Видео:Хроматэк Аналитик 3.1. Создание метода с градуировкойСкачать

РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют 4 основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
Метод нормирования (метод внутренней нормализации).
Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:

 – сумма площадей (высот) всех пиков на хроматограмме.
– сумма площадей (высот) всех пиков на хроматограмме.
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.
Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле:

Сi и С0 – концентрация i-го вещества и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-го вещества и стандартного вещества соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8 – 1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:

При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8 – 1,2.
Метод внешнего стандарта.
Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах раствора стандартного образца.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:

S и S0 – средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов соответственно;
С и С0 – концентрации определяемого и стандартного растворов соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве раствора стандартного образца для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в растворе стандартного образца по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8 — 1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой, в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
Метод внутреннего стандарта.
Концентрацию определяемого вещества определяют путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме раствора стандартного образца. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле:

где  – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
 – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в растворе стандартного образца;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в растворе стандартного образца;
С0 – концентрация определяемого вещества в растворе стандартного образца.
В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
Метод стандартных добавок.
Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условиях с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:

Сстд — концентрация стандартной добавки;
Sx — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
Видео:Нормальное Распределение за 6 МинутСкачать

РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
Если не указано иное, в испытаниях на содержание примесей в качестве порога игнорирования (неучитываемый предел, уровень содержания вещества, при котором и при меньшем содержании пик не принимается во внимание) принимают величину 0,05 % от содержания основного вещества.
В случаях, когда при испытании на примеси требуется определение суммарного содержания примесей или количественного определения примеси, при интерпретации хроматограммы важно выбрать подходящие условия интегрирования и значения порога интегрирования [интенсивность сигнала пика соединения (высоты или площади)], при котором (и при меньшем) пик не учитывается системой сбора данных и не участвует в количественных расчетах.
С целью разметки на хроматограмме всех пиков, подлежащих учету, заданный порог интегрирования системы сбора данных не должен превышать половину порога игнорирования.
Интегрирование площади пика любой примеси, пик которой не полностью разделяется с основным пиком, предпочтительно проводить экстраполяцией смещенной базовой линии (методом тангенциальной касательной). Использование других способов интегрирования должно быть специально оговорено в фармакопейной статье.
Видео:Ошибки при построении градуировкиСкачать

ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
Испытания пригодности системы являются неотъемлемой частью методики и используются для того, чтобы убедиться в надлежащем функционировании хроматографической системы и обеспечить выполнение предъявляемых к ней требований.
При проведении испытаний используемое оборудование должно быть квалифицировано и способно к функционированию надлежащим образом.
Для оценки пригодности системы указывают:
- требования к параметрам, характеризующим форму пика [эффективность хроматографической системы N и фактор асимметрии (фактор симметрии) As];
- требования к разделительной способности (разрешение между пиками RS или отношение пик — долина p/v);
- требования к воспроизводимости (относительное стандартное отклонение, RSD) значений площади или высоты пиков, а также времен удерживания пиков в случае оценки подлинности соединений;
- требования к чувствительности при проведении испытания на примеси [минимально определяемая концентрация (фактический предел количественного определения) примеси]. Оценивается посредством расчета отношения сигнал/шум для раствора соответствующей концентрации.
Если в фармакопейной статье не указано иное, должны выполняться следующие требования и все дополнительные требования, приведенные в нормативном документе:
- в испытаниях на примеси или количественное содержание для пика на хроматограмме растворе стандартного образца, используемого для количественных определений, значение величины фактора асимметрии (фактора симметрии) As должно находиться в пределах от 0,8 до 1,5;
- разрешение между пиками Rs ³ 1,5 (в случае не полностью разделенных пиков вместо разрешения может быть использовано соотношение p/v. При этом требования к минимальному значению соотношения p/v устанавливаются в фармакопейной статье);
- отношение сигнал/шум для пика вещества, полученное для раствора с концентрацией, равной требуемому уровню минимально определяемой концентрации, должно быть не менее 10. Требуемый уровень минимально определяемой концентрации [фактический предел количественного определения (ПКО)] зависит от того, предполагает ли методика вычисление содержания примесей, или только полуколичественную оценку, когда результат представляется в виде «менее Х» или «не более Х», где Х – допустимое содержание примеси. Для методик, предполагающих вычисление содержания примесей, минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать значение порога игнорирования (если не указано иное – 0,05 % относительно концентрации основного вещества в испытуемом растворе). Для полуколичественных методик минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать максимально допустимое содержание примеси. При необходимости оценки содержания нескольких примесей требуемый уровень минимальной определяемой концентрация для применяемой хроматографической системы определяется примесью, нормы содержания которой наиболее строги. Если в фармакопейной статье не указано иное, то раствор вещества минимально определяемой концентрации для оценки чувствительности детектирования можно приготовить растворением стандартного образца вещества в том же растворителе, который используется для приготовления испытуемого раствора, с уровнем концентрации 0,05 % относительно концентрации основного вещества в испытуемом растворе.
Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
При количественном определении основного вещества в фармацевтических субстанциях при содержании основного вещества, близком к 100 %, максимально допустимое относительное стандартное отклонение значений интенсивности (площади или высоты) пика основного вещества для заданного количества повторных введений стандартного раствора RSDmax вычисляется по формуле:

где К – константа (0,349), полученная из выражения:

в котором множитель  соответствует значению RSDmax после 6 повторных инжекций для В = 1,0;
соответствует значению RSDmax после 6 повторных инжекций для В = 1,0;
В – верхний предел содержания основного вещества, приведенный в фармакопейной статье, минус 100 %;
n – число повторных введений стандартного раствора, (3 ≤ n ≤ 6);
t90 %, n‑1 – коэффициент Стьюдента (двусторонний) при доверительной вероятности 90 % с (n – 1) степенью свободы.
Если в фармакопейной статье не указано иное, то значение RSD не должно превышать величин RSDmax, приведенных в таблице. Данное требование не применяется при испытаниях на примеси.
Таблица – Максимально допустимое относительное стандартное отклонение RSDmax в зависимости от верхнего предела содержания основного вещества и числа отдельных введений проб
| В, % | Число отдельных введений проб (вколов) | |||
| 3 | 4 | 5 | 6 | |
| Максимально допустимое относительное стандартное отклонение RSDmax | ||||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
Соответствие критериям пригодности системы должно поддерживаться на протяжении всего испытания. В зависимости от различных факторов, например, частоты использования методики и опыта работы с хроматографической системой, аналитик выбирает соответствующую схему проверки, подтверждающую соответствие этим требованиям.
В планарной хроматографии при проведении испытаний, регламентирующих наличие посторонних примесей/родственных соединений, должно быть предусмотрено определение предела обнаружения определяемых примесей и разделительной способности хроматографической системы.
Для подтверждения пригодности системы возможно одновременное использование двух тестов: «Подтверждение разделительной способности» и «Подтверждение чувствительности».
Тест «Подтверждение разделительной способности» проводят путём нанесения на пластинку и последующего хроматографирования специального стандартного раствора, содержащего два или более веществ с известными значениями Rf. После хроматографирования эти вещества должны разделиться, образуя зоны адсорбции, значения Rf которых равны заданным.
Тест «Подтверждение чувствительности» позволяет оценить чувствительность проявления. Он заключается в нанесении определённого количества стандартного вещества, хроматографируемого в условиях предыдущего теста и проявляемого тем же реагентом. Зона адсорбции стандартного вещества должно чётко обнаруживаться.
В каждом случае природа стандартных веществ, состав стандартного раствора, наносимые количества, приблизительные значения Rf устанавливают в фармакопейных статьях.
При необходимости для достижения требуемых критериев пригодности хроматографической системы проводится корректировка хроматографических условий.
Видео:Курс Interlab "Хроматография: газовая хроматография. Детекторы." Лекция 5Скачать

КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
Далее приводятся пределы возможных изменений различных параметров хроматографической методики, которые могут быть внесены в нее для соответствия требованиям пригодности системы без принципиального изменения методик. Корректировка условий градиентного элюирования является более критичной, чем изократических условий, так как может вызвать сдвиг пиков на другую стадию градиента, и, таким образом, привести к некорректной идентификации пиков, наложению пиков или сдвигов, при которых элюирование интересующих соединений происходит после указанного времени регистрации хроматограммы. При внесении изменений, отличных от приведенных ниже, требуется проведение повторной валидации методики.
Проверка пригодности системы должна быть включена в методики для подтверждения получения разделения, требуемого для надлежащего проведения испытания. Тем не менее, поскольку неподвижные фазы описаны только в общем виде, и существует огромное количество доступных коммерческих фаз, отличающихся по хроматографическому поведению, некоторая корректировка условий хроматографирования может потребоваться для выполнения требований пригодности системы. В методиках с использованием обращенно-фазовой жидкостной хроматографии корректировки различных параметров не всегда приводят к удовлетворительному разделению. В этом случае может возникнуть необходимость замены одной колонки на другую, такого же типа, обеспечивающую желаемое хроматографическое поведение.
Если в фармакопейной статье не указано иное, допустимы следующие изменения параметров хроматографической системы.
Тонкослойная и бумажная хроматография
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
Пример 1: для вещества, содержание которого составляет 10 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 7 – 13 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению абсолютного содержания до 8 – 12 %. Допустимое изменение по относительному содержанию больше, чем допустимое изменение по абсолютному содержанию. Таким образом, допускается изменение содержания 7 – 13%.
Пример 2: для вещества, содержание которого составляет 5 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 3,5 – 6,5 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению его абсолютного содержания в подвижной фазе до 3 – 7 %. В этом примере допустимое изменение по абсолютному содержанию больше, чем допустимое изменение по относительному содержанию, и допустимым диапазоном содержания является 3 – 7 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Наносимый объём. При использовании пластинок с малым размером частиц сорбента (2 — 10 мкм) наносимый объём может быть изменен на 10 –20 % от заявленного объёма.
Жидкостная хроматография: изократическое элюирование
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Скорость потока подвижной фазы: ± 50 %, большая степень изменений допустима при одновременном изменении размеров колонки. При необходимости одновременного изменения размеров колонки и скорости потока сначала рассчитывается номинальная скорость потока для колонки, размеры которой отличаются от приведенной в нормативном документе, а затем допускается корректировка полученного значения скорости потока на ± 50 %.
- замена типа неподвижной фазы недопустима (например, недопустима замена фазы С18 на фазу С8);
- размер частиц: максимальное уменьшение размера частиц 50 %, увеличение не допускается.
Длина колонки: ± 70 %,
внутренний диаметр колонки: ± 25 %.
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

F1 — скорость потока, указанная в нормативном документе, мл/мин;
F2 — скорректированная скорость потока, мл/мин;
l1 — длина колонки, указанная в нормативном документе, мм;
d1 — внутренний диаметр колонки, указанный в нормативном документе, мм;
d2 — внутренний диаметр используемой колонки, мм.
Температура: ± 10 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы (минимально определяемая концентрация) и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Жидкостная хроматография: градиентное элюирование
Изменение хроматографических условий для систем с градиентом требует большей осторожности, чем для изократических.
Состав подвижной фазы/профиль градиентного элюирования: незначительные изменения состава подвижной фазы и системы градиента являются приемлемыми при условии, что:
— выполнены требования пригодности системы;
— основной пик элюируется в пределах ± 15 % от обозначенного времени удерживания;
— элюирующая способность конечного состава подвижной фазы не менее предписанного состава.
Если при использовании градиентного элюирования не может быть достигнуто соответствие требованиям пригодности системы, рекомендуется оценить объем задержки градиента или сменить используемую колонку.
Объем задержки градиента. Конфигурация используемого оборудования может значительно изменить разрешение, указанные абсолютные и относительные времена удерживания. Это может произойти из-за отличия объема задержки градиента используемой системы от объема задержки градиента системы, с помощью которой проводилась валидация методики. В нормативном документе часто перед началом программы градиента включена изократическая стадия, чтобы можно было адаптировать систему к временным точкам градиента с учетом разницы объема задержки у системы, использованной для разработки методики, и у фактически использующейся системы. Решение о необходимости адаптации длительности изократической стадии для данного аналитического оборудования принимается пользователем. Если объем задержки, использованный при разработке методики, приведен в нормативном документе, то интервалы времени (t), указанные в таблице градиента, можно заменить адаптированными интервалами времени (tc), рассчитанными с помощью следующего уравнения:

где D – объем задержки, мл;
D0 – объем задержки, использованный при разработке методики, мл;
F – скорость потока, мл/мин.
Изократическая стадия, введенная с этой целью, может быть исключена, если имеются данные валидации по применению методики без этой стадии.
pH среды водного компонента подвижной фазы. Изменения не допускаются.
Концентрация солей в буферном веществе подвижной фазы. Изменения не допускаются.
Скорость потока. Изменения допустимы при изменении размеров колонки.
— замена типа неподвижной фазы недопустима (например, недопустима замена С18 на С8);
— размер частиц: изменения не допускаются;
— длина колонки: ± 70 %;
— внутренний диаметр колонки: ± 25 %;
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

где F1 — скорость потока, указанная в нормативном документе, мл/мин;
F2 — скорректированная скорость потока, мл/мин;
l1 — длина колонки, указанная в нормативном документе, мм;
d1— внутренний диаметр колонки, указанный в нормативном документе мм;
d2 — внутренний диаметр используемой колонки, мм.
Температура. ± 5 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что детектирование (предел количественного определения) и сходимость отклика (RSD площадей или высот) для пика (пиков) определяемых соединений остаются удовлетворительными.
Газовая хроматография
— размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки);
— толщина плёнки: от минус 50 % до плюс 100 % (капиллярные колонки).
-Длина колонки: ± 70 %;
-Внутренний диаметр колонки: ± 50 %.
Скорость потока: ± 50 %.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Сверхкритическая флюидная хроматография
Состав подвижной фазы. Для набивных колонок количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше; для систем с капиллярной колонкой изменения не допускаются.
Длина волны детектора. Изменения не допускаются.
— размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки).
— Длина колонки: ± 70 %;
— Внутренний диаметр колонки: ± 25 % (набивные колонки), ± 50 % (капиллярные колонки).
Скорость потока: ± 50 %.
Температура: ± 5 %, если температура указана в нормативном документе.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Видео:Решение задачи на построение калибровочного графика и расчет концентрации аналита в образце (excel)Скачать
Хроматографические методы анализа. Учебно-методическое пособие (стр. 3 )
 | Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 4 5 |

Этот простой и точный метод является основным методом определения микропримесей. Кроме того, метод не требует разделения всех компонентов смеси, а ограничивается лишь теми, определение которых необходимо в данном конкретном случае.
Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандартного вещества. В качестве стандартного выбирают вещество, близкое по физико-химическим свойствам к компонентам смеси. Это вещество должно отсутствовать в исследуемой смеси и давать на хроматограмме пик, отдельный от других компонентов. После хроматографирования измеряют площади пиков анализируемого компонента (Si) и стандартного вещества (SCT).Массовую долю компонента (Wi, %) рассчитывают по формуле:
 (1.17),
(1.17),
где r – отношение массы внутреннего стандарта к массе пробы.
Достоинством метода внутреннего стандарта является хорошая воспроизводимость, высокая точность, отсутствие влияния на измеряемые величины небольших колебаний условий опыта.
К недостаткам относятся требование точной дозировки стандарта и хорошего отделения пика стандарта от пиков анализируемых веществ. Пользование калибровкой возможно только для той области концентраций, в которой сохраняется линейная зависимость между показаниями детектора и концентрацией определяемого вещества.
Метод простой нормировки чаще всего используют на практике. Для его использования необходимо, чтобы на хроматограмме были зарегистрированы все компоненты, входящие в состав анализируемой смеси; сумму площадей всех пиков принимают за 100 %. Тогда отношение площади одного пика к сумме площадей, умноженное на 100, будет характеризовать массовую долю (%) компонента в смеси.
Этот метод основан на том предположении, что вещества, взятые в одинаковом количестве, дают одну и ту же площадь пика, независимо от их строения. Это приближенно выполняется, если вещества химически сходны, а в качестве газа-носителя применяется газ с высокой теплопроводностью (водород или гелий).
Если чувствительность детектора различна по отношению к разделяемым компонентам смеси, то используют метод нормировки с поправочными коэффициентами. В этом случае расчет ведут по формуле (1.18), где ki – поправочный коэффициент i-го компонента (мг/см2):
 (1.18)
(1.18)
Поправочные коэффициенты получают при анализе стандартных серий и рассчитывают по формуле
 (1.19)
(1.19)
где С – концентрации определяемого и стандартных веществ. Метод нормировки требует полного разделения и идентификации всех компонентов смеси; необходимости в знании калибровочных коэффициентов для всех без исключения компонентов смеси. Ошибки в определении параметра пика или калибровочного коэффициента какого-либо одного компонента приводят к неверным результатам всего анализа. Поэтому метод нормировки применяется главным образом для рутинных анализов малокомпонентных смесей и для приближенных результатов.
5.8. Практическое применение газовой хроматографии
Газовая хроматография – один из наиболее перспективных физико-химических методов анализа. В настоящее время вряд ли существует научно-исследовательская или производственная лаборатория, занимающаяся анализом органических веществ, в которой отсутствовала бы хроматографическая аппаратура.
Методом газовой хроматографии анализируют нефтяные и рудничные газы, воздух, продукцию основной химии и промышленности основного органического синтеза, нефть и продукты ее переработки, металлоорганические соединения и т. д. Газовая хроматография используется в биологии, медицине, в технологии переработки древесины, в лесохимии и пищевой промышленности.
Выпускаемая аппаратура позволяет анализировать не только вещества, представляющие собой в нормальных условиях газы, но и высококипящие соединения, фармацевтические препараты, различные пестициды и т. д.
Газовая хроматография применяется также для автоматизации производственных процессов. Датчик промышленного хроматографа используется не только как регистрирующий прибор, но и как регулирующее устройство, подающее сигналы непосредственно исполнительным механизмам. Таким образом, промышленный хроматограф может контролировать и регулировать важнейшие параметры технологического процесса: температуру, давление, расход сырья и т. д.
Из физико-химических применений газовой хроматографии следует отметить возможность изучения термодинамики сорбции, определения молекулярных масс, давления пара веществ, коэффициентов диффузии, поверхности адсорбентов и катализаторов.
Важной особенностью газовой хроматографии является возможность определения в различных продуктах микропримесей. В настоящее время методом газовой хроматографии удается определять концентрации порядка 10-10%. Это делает метод незаменимым при анализе мономеров, используемых в производстве полимерных материалов, а также при исследовании биосферы.
Вопросы для самоконтроля
1. Что такое: а) общее время удерживания; б) приведенное (исправленное) время удерживания; в) общий удерживаемый объем; г) приведенный (исправленный) удерживаемый объем?
2. В чем сущность качественного хроматографического анализа по величине удерживаемого объема?
3. Почему предпочитают использовать исправленный удерживаемый объем, а не объем удерживания?
4. Какие хроматографические параметры можно использовать для идентификации компонентов смеси?
5. Почему идентификацию компонентов не рекомендуется вести по абсолютным временам удерживания?
6. Какие другие способы идентификации компонентов применяют в хроматографическом анализе?
7. Можно ли сделать вывод о природе веществ на основании хроматографических данных?
8. Как зависит время (объем) удерживания от растворимости соединения в подвижной фазе?
9. Что такое относительный удерживаемый объем и относительное время удерживания?
10. В чем сущность основных методов количественной хроматографии: а) нормировки с поправочными коэффициентами; б) абсолютной калибровки; в) внутреннего стандарта?
11. Укажите возможности и ограничения различных количественных методов хроматографического анализа.
12. Как можно измерить площадь пика на хроматограмме? Какой зависимостью связана площадь пика с концентрацией вещества?
13. Почему способ абсолютной калибровки сравнительно редко применяют в хроматографических лабораториях?
14. Как повысить точность определения компонента по методу нормировки?
15. Какие параметры хроматографичеcкого пика используют для количественного анализа?
16. В каких случаях в количественном хроматографическом анализеизмеряют высоту пика? площадь пика?
6. Жидкостная колоночная хроматография
Жидкостная хроматография (ЖХ) – это метод разделения и анализа сложных смесей веществ, в котором подвижной фазой служит жидкость. Метод ЖХ применим для разделения более широкого круга веществ, чем метод ГХ, поскольку большинство веществ не обладает летучестью, многие из них неустойчивы при высоких температурах. В ЖХ разделение чаще всего происходит при комнатной температуре. Жидкая подвижная фаза, в отличие от газа в ГХ, выполняющего только транспортную функцию, является активным элюентом. Молекулы жидкой фазы могут сорбироваться на поверхности неподвижной фазы. При прохождении через колонку находящиеся в элюенте молекулы интересуюшего нас компонента должны вытеснить молекулы элюента с поверхности сорбента. Применяя различные элюенты, можно изменять параметры удерживания и селективность хроматографической системы.
В классическом варианте ЖХ в стеклянную колонку длиной 1–2 м, заполненную сорбентом (размер частиц  100 мкм), вводят анализируемую пробу и пропускают элюент. Скорость прохождения элюента под действием силы тяжести мала, а продолжительность анализа значительна. Однако такой вариант ЖХ не требует дорогостоящего оборудования и до сих пор находит применение.
100 мкм), вводят анализируемую пробу и пропускают элюент. Скорость прохождения элюента под действием силы тяжести мала, а продолжительность анализа значительна. Однако такой вариант ЖХ не требует дорогостоящего оборудования и до сих пор находит применение.
Вследствие использования сорбентов со значительно меньшим размером частиц (до 5–10 мкм), нагнетательных насосов, чувствительных детекторов произошел переход от классической к высокоэффективной жидкостной хроматографии (ВЭЖХ), позволяющей проводить разделение и определение молекул, ионов, разделение макромолекул и биологически активных молекул. К достоинствам метода ВЭЖХ можно отнести универсальность, возможность автоматизации разделения и анализа сложных смесей органических и неорганических веществ, экспрессность, эффективность и высокую чувствительность. Это серийный метод определения органических соединений многих классов, его широко используют при анализе смесей аминокислот, белков, лекарственных препаратов. ВЭЖХ находит применение и в неорганическом анализе для разделения ионов в зависимости от их размера.
6.1. Адсорбционная хроматография
В адсорбционном варианте жидкостной хроматографии в зависимости от полярности неподвижной и подвижной фаз различают нормально-фазовую (НФХ) и обращенно-фазовую (ОФХ) хроматографии. В НФХ используют полярный адсорбент и неполярные подвижные фазы. В ОФХ – неполярный адсорбент и полярные подвижные фазы. Неподвижная фаза должна удерживать разделяемые компоненты. Подвижная фаза, т. е.растворитель, должна обеспечить различную емкость колонки и эффективное разделение за приемлемое время.
Неподвижные фазы. В качестве адсорбентов применяют тонкодисперсные пористые материалы.
Полярные адсорбенты ( ,
,  , оксиды металлов, флорисил и др.) имеют на поверхности слабокислотные ОН-группы, способные удерживать вещества с основными свойствами. Недостаток полярных сорбентов – высокая чувствительность к содержанию воды в растворителях, приводящая к изменению свойств поверхности и невоспроизводимым результатам анализа. Для ВЭЖХ применяют полярные сорбенты с привитыми полярными группами (амины, диолы и др.), что позволяет менять селективность, подбирая подходящий элюент.
, оксиды металлов, флорисил и др.) имеют на поверхности слабокислотные ОН-группы, способные удерживать вещества с основными свойствами. Недостаток полярных сорбентов – высокая чувствительность к содержанию воды в растворителях, приводящая к изменению свойств поверхности и невоспроизводимым результатам анализа. Для ВЭЖХ применяют полярные сорбенты с привитыми полярными группами (амины, диолы и др.), что позволяет менять селективность, подбирая подходящий элюент.
Неполярные адсорбенты (графитированная сажа, кизельгур, диатомит) не проявляют селективности к полярным молекулам. Используют также сорбенты с привитыми неполярными фазами, например силикагель с алкилсилильными группами от С2 до С22.
Подвижные фазы. В ЖХ важен выбор подвижной фазы, поскольку она оказывает большое влияние на селективность разделения, эффективность колонки и скорость движения хроматографической полосы. Подвижная фаза должна растворять анализируемую пробу, обладать малой вязкостью, из нее должно быть возможным выделение разделенных компонентов. Подвижная фаза должна быть инертна по отношению к материалам всех частей хроматографа, безопасной, дешевой.
Разделение компонентов достигают, меняя элюирующую силу растворителя.
Элюирующая сила растворителя показывает, во сколько раз энергия сорбции данного элюента больше, чем энергия сорбции элюента, выбранного в качестве стандарта, например н-гептана.
Растворители (элюенты) делят на слабые и сильные. Слабые растворители слабо адсорбируются неподвижной фазой, поэтому коэффициенты распределения сорбируемых веществ (сорбата) высокие. Сильные растворители сильно адсорбируются, поэтому коэффициенты распределения сорбата низкие. Растворитель тем сильнее, чем выше растворимость в нем анализируемой пробы, чем сильнее взаимодействие растворитель–сорбат.
Элюирующая сила определяется полярностью растворителя. В НФХ с увеличением полярности растворителя элюирующая сила растворителя растет, в ОФХ – снижается. Часто применяют не индивидуальные растворители, а их смеси. Незначительные добавки другого растворителя, особенно воды, существенно увеличивают элюирующую силу элюента.
При разделении многокомпонентных смесей одна подвижная фаза в качестве элюента может не разделить все компоненты пробы. В этом случае применяют метод ступенчатого или градиентного элюирования, применяя в процессе хроматографирования последовательно все более сильные элюенты. Установлены некоторые эмпирические правила, помогающие при выборе элюента. Сорбция, как правило, увеличивается с ростом числа двойных связей и ОН-групп в соединениях. Сорбция уменьшается в ряду органических соединений: кислоты–спирты–альдегиды–кетоны–сложные эфиры–ненасыщенные углеводороды–насыщенные углеводороды.
Для разделения веществ разной полярности и для разделения соединений разных классов применяют НФХ. В ОФХ неподвижная фаза сильнее адсорбирует неполярные компоненты из полярных элюентов, например из воды.
Метод адсорбционной ВЭЖХ – это серийный метод определения органических соединений многих классов, его широко используют при анализе смесей аминокислот, белков, лекарственных, препаратов.
6.2. Распределительная хроматография.
Метод распределительной, или жидкостно-жидкостной, хроматографии основан на распределении вещества между двумя несмешивающимися жидкостями, подобно тому, как это происходит в многократной ступенчатой экстракции. Жидкую неподвижную фазу наносят на пористый достаточно инертный сорбент и заполняют им распределительную колонку. При пропускании жидкой подвижной фазы через колонку смесь разделяется на компоненты главным образом за счет их различной растворимости в жидкой неподвижной фазе. Обычно растворимость компонентов пробы в подвижной и неподвижной фазах, обладающих разной полярностью, сильно различается. Если растворимость пробы выше в неподвижной фазе, то время удерживания компонентов значительно возрастает. Если растворимость пробы выше в подвижной фазе, то время удерживания может быть близким к времени удерживания несорбируемого компонента. Чтобы добиться разделения, в подвижную фазу, насыщенную неподвижной, включают третий компонент, снижающий различие в полярности подвижной и неподвижной фаз. Например, к смеси из неполярного (гексан) и полярного (вода) растворителей прибавляется спирт.
В нормально-фазовой распределительной хроматографии используют следующие системы: полярный растворитель (вода, спирт) фиксирован на твердом носителе – силикагеле, диатомите, целлюлозе, оксиде алюминия. Полярной фазой в этом случае служат неполярные растворители – изооктан, бензол, и др.
В обращенно-фазовой распределительной хроматографии неполярный растворитель фиксируют на носителе, а в качестве подвижной фазы используют полярные растворители (вода, спирт, буферные растворы, сильные кислоты).
Нанесенные жидкие фазы имеют большой недостаток – они быстро смываются подвижной жидкой фазой с поверхности носителя, особенно, при использовании таких систем в ВЭЖХ, т. е. при повышенном давлении в колонке. Поэтому жидкие фазы прививают к носителю. В качестве носителей неподвижных жидких фаз для НФРХ используют силикагели с привитыми нитрильными, аминными и другими группами. В обращенно-фазовом варианте используют силикагели с привитыми алкилсилильными группами. Механизм удерживания на таких сорбентах сложен.
Метод распределительной хроматографии применяют для разделения сильнополярных соединений, аминокислот, фенолов, фенилкарбоновых кислот и др.
6.3. Эксклюзионная хроматография
Эксклюзионная хроматография – это разновидность жидкостной хроматографии, в которой разделение компонентов основано на распределении молекул в соответствии с их размером между растворителем, находящимся в порах сорбента, и растворителем, протекающим между его частицами. В процессе разделения небольшие молекулы попадают в сетку полимера, в порах которой растворитель служит неподвижной фазой, и удерживаются там, большие молекулы не могут проникнуть в полимерную сетку и вымываются из колонки подвижной фазой. Вначале элюируются самые большие, затем средние и потом небольшие молекулы. Поэтому эксклюзионную хроматографию называют также молекулярно-ситовой. Эксклюзионная хроматография подразделяется на гель-проникающую и гель-фильтрационную. В гель-проникающей хроматографии разделение осуществляется на полимерах, набухающих в органических растворителях; если же полимеры набухают в воде, то говорят о гель-фильтрационном варианте.
Каждый сорбент характеризуется объемом пор, следовательно, областью разделяемых молекулярных масс и градуировочным графиком, который имеет сложный вид, характеризующий зависимость удерживаемого объема от молекулярной массы или размера молекул. Надо подбирать сорбент и длину хроматографической колонки такими, чтобы разделение вещества протекало в пределах линейного участка градуировочного графика.
Неподвижные фазы в эксклюзионной хроматографии выбирают для конкретной аналитической задачи. Первоначально устанавливают, какая система растворителей может быть использована для анализа (водная или водно-органическая), что и определяет тип сорбента.
Подвижные фазы в эксклюзионной хроматографии должны удовлетворять определенным требованиям:
– полное растворение образца;
– хорошее смачивание сорбента;
– низкая вязкость и токсичность.
Метод эксклюзионной хроматографии широко используют при исследовании полимеров, определении их молекулярных масс, а также в биологии и медицине для анализа белков, крови и других объектов.
6.4. Особенности жидкостных хроматографов.
Жидкостной хроматограф – более сложный прибор по сравнению с газовым. Это связано с тем, что система подачи элюента включает ряд дополнительных узлов: систему дегазации, градиентное устройство, насосы и измерители давления.
Градиентное устройство должно обеспечить отбор элюентов из двух-трех емкостей в смеситель, затем в колонку. Насосы должны иметь постоянную скорость потока от 0.1 до 10 мл/мин при давлении 400 атм. Кроме того, необходимо тщательное удаление газа из всех используемых растворителей, так как появление пузырьков газа в детекторе недопустимо. Проба вводится с помощью петлевых дозаторов или специальных микрошприцов через прокладку из специальных ненабухающих полимерных материалов.
В ВЭЖХ обычно используют прямые колонки длиной 10, 15, 25 см с внутренним диаметром 4–5,5 мм. В микроколочных хроматографах используют колонки длиной 5–6 см и диаметром 1–2 мм. Колонки изготавливают из стекла или нержавеющей стали.
В ионном хроматографе все соединительные трубки, колонки, краны выполнены из химически инертных материалов, что позволяет использовать сильнокислотные и сильноосновные элюенты.
Для непрерывного контроля элюата, вытекающего из колонки, обычно используют дифференциальные рефрактометры, люминесцентные, УФ-спектрофотометрические и кондуктометрические детекторы.
Дифференциальный рефрактометр – это универсальный детектор, который позволяет определять общий показатель преломления системы проба– элюент, т. е. сигнал дают все компоненты, показатель преломления которых отличается от показателя преломления элюента. Чувствительность детектора – 10-6 г.
УФ-детектор работает при одной и той же длине волны, соответствующей наиболее интенсивной линии ртутной лампы низкого давления  =253.7 нм. УФ-детектор наиболее чувствителен, если молярные коэффициенты светопоглощения компонентов высоки, а элюент не поглощает в ультрафиолетовой области спектра. С помощью такого детектора можно определять любые ароматические соединения, большинство кетонов и альдегидов. УФ-детектор селективен, чувствительность его составляет 10-9 г.
=253.7 нм. УФ-детектор наиболее чувствителен, если молярные коэффициенты светопоглощения компонентов высоки, а элюент не поглощает в ультрафиолетовой области спектра. С помощью такого детектора можно определять любые ароматические соединения, большинство кетонов и альдегидов. УФ-детектор селективен, чувствительность его составляет 10-9 г.
Фотометры и спектрофотометры позволяют работать при любой длине волны (190-650 нм). Регистрируют изменение поглощения во времени при определенной длине волны или в остановленном потоке элюента снимают спектр. Быстрозаписывающий спектрофотометр позволяет записать всю спектральную область за 20с.
Кондуктометрический детектор применяют в ионной хроматографии для измерения проводимости раствора, пропорциональной числу ионов в растворе, их подвижности. Предел обнаружения с помощью такого детектора составляет порядка 10-3 мкг/мл. Использование концентрирующей колонки позволяет снизить предел обнаружения на 2–3 порядка.
В эксклюзионной хроматографии при анализе полимеров для определения средних молекулярных масс используют проточный нефелометр.
Вопросы для самоконтроля
1. Какова роль подвижной фазы в жидкостной хроматографии?
2. Какими способами проба анализируемой смеси веществ вводится в хроматографическую установку в жидкостной хроматографии?
3. Какова роль основных узлов в жидкостном хроматографе высокого давления? Что общего и каковы принципиальные отличия от газового хроматографа?
4. Назовите три способы детектирования в жидкостной хроматографии.
5. Почему в жидкостной хроматографии предпочитают подвижные фазы с низкой вязкостью?
6. Какие варианты используются в жидкостно-жидкостной распределительной хроматографии?
7. Каковы особенности эксклюзионной хроматографии?
8. Как изменяется время (объем) удерживания молекул в эксклюзионной хроматографии с увеличением их размера?
9. Чем отличаются нормально — и обращенно-фазовый варианты ВЭЖХ?
ГЛАВА 2. ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1. Определение качественного состава смеси на основе
Цель работы: Идентифицировать компоненты хроматографируемой смеси по объемам удерживания и для идентифицированных компонентов рассчитать удельные удерживаемые объемы.
Cущность работы: Газовая хроматография позволяет проводить индивидуальную и групповую идентификацию веществ (т. е. отнесение их к определенной группе соединений). Индивидуальную хроматографическую идентификацию проводят с помощью следующего приема: сравнивают характеристики удерживания компонентов анализируемой смеси с характеристиками удерживания стандартов, компонентов стандартных смесей или с табличными данными.
Видео:Курс Interlab "Хроматография: газовая хроматография. Ввод пробы. Часть 2." Лекция 7Скачать

Характеристики удерживания
1. Расстояние удерживания  , мм – расстояние от момента ввода пробы на хроматограмме до максимума соответствующего пика.
, мм – расстояние от момента ввода пробы на хроматограмме до максимума соответствующего пика.  – расстояние удерживания несорбирующегося компонента, мм, определяется от момента ввода пробы до максимума пика несорбирующегося компонента;
– расстояние удерживания несорбирующегося компонента, мм, определяется от момента ввода пробы до максимума пика несорбирующегося компонента;
2. Исправленное (приведенное) расстояние удерживания,  – расстояние от вершины пика несорбирующегося компонента до максимума соответствующего пика, мм;
– расстояние от вершины пика несорбирующегося компонента до максимума соответствующего пика, мм;
 (2.1)
(2.1)
3. Время удерживания  ,мин – время, прошедшее от момента ввода пробы в колонку до момента записи максимума соответствующего пика.
,мин – время, прошедшее от момента ввода пробы в колонку до момента записи максимума соответствующего пика.
Время удерживания можно рассчитать, зная линейную скорость движения диаграммной ленты в потенциометре Uл, мм/мин:
 (2.2)
(2.2)
(часто время удерживания измеряется непосредственно с помощью секундомера).

Рис. 2.1. Хроматограмма, иллюстрирующая расстояние удерживания
4. Исправленное время удерживания 
 , мин – время, прошедшее с момента появления максимума пика несорбирующегося компонента до появления максимума пика соответствующего соединения,
, мин – время, прошедшее с момента появления максимума пика несорбирующегося компонента до появления максимума пика соответствующего соединения,
 (2.3),
(2.3),
где  – время удерживания несорбирующегося компонента.
– время удерживания несорбирующегося компонента.
Время удерживания есть функция длины колонки, скорости потока газа-носителя, сорбируемости, температуры. Величиной, не зависящей от скорости потока газа-носителя, является объем удерживания.
5. Объем удерживания (удерживаемый объем),  , мл – объем газа- носителя при температуре колонки, прошедшего через колонку за время от момента ввода пробы до момента регистрации максимума соответствующего пика хроматограммы.
, мл – объем газа- носителя при температуре колонки, прошедшего через колонку за время от момента ввода пробы до момента регистрации максимума соответствующего пика хроматограммы.
 (2.4),
(2.4),
где  –исправленная объемная скорость газа носителя, мл/мин.
–исправленная объемная скорость газа носителя, мл/мин.
Объемная скорость газа-носителя  измеряется на выходе из колонки жидкостным расходомером. Для расчета истинного (исправленного)значения, , следует учитывать давление насыщенного пара рабочей жидкости расходомера (обычно воды),
измеряется на выходе из колонки жидкостным расходомером. Для расчета истинного (исправленного)значения, , следует учитывать давление насыщенного пара рабочей жидкости расходомера (обычно воды),  , при температуре расходомера
, при температуре расходомера  , тогда,
, тогда,
 (2.5)
(2.5)
где — измеренная объемная скорость расхода газа-носителя на выходе из колонки, мл/мин.;  — давление газа на выходе из колонки, обычно 1 атм;
— давление газа на выходе из колонки, обычно 1 атм;  – температура колонки, К.
– температура колонки, К.
6. Приведенный (исправленный) объем удерживания  , мл – объем удерживания с поправкой на объем удерживания несорбирующегося компонента (
, мл – объем удерживания с поправкой на объем удерживания несорбирующегося компонента ( ). Рассчитывается по формуле:
). Рассчитывается по формуле:
 (2.6)
(2.6)
7. Эффективный (истинный) объем удерживания  ,мл – приведенный объем удерживания, исправленный с учетом перепада давления в колонке:
,мл – приведенный объем удерживания, исправленный с учетом перепада давления в колонке:
 (2.7),
(2.7),
где j – поправочный коэффициент, учитывающий сжимаемость газа носителя в колонке:
 (2.8)
(2.8)
где  – давление газа-носителя на входе в колонку, атм;
– давление газа-носителя на входе в колонку, атм;  – давление газа-носителя на выходе из колонки, равное 1 атм.
– давление газа-носителя на выходе из колонки, равное 1 атм.
Таким образом, истинный удерживаемый объем равен:
 (2.9)
(2.9)
6. Удельный объем удерживания при температуре колонки,  – истинный объем удерживания, отнесенный к единице массы неподвижной жидкой фазыв колонке. Рассчитывается по формуле:
– истинный объем удерживания, отнесенный к единице массы неподвижной жидкой фазыв колонке. Рассчитывается по формуле:
 (2.10),
(2.10),
где q – масса неподвижной жидкой фазы в колонке, г.
9. Удельный объем удерживания  – удельный объем удерживания при температуре колонки, приведенный к 273 К, рассчитывается по формуле:
– удельный объем удерживания при температуре колонки, приведенный к 273 К, рассчитывается по формуле:
 (2.11)
(2.11)
В газожидкостной хроматографии удельный удерживаемый объем представляет собой физико-химическую константу. Значение ее зависит только от природы жидкой фазы и не зависит от условий хроматографического разделения. Величина может быть использована для идентификации компонентов в качественном анализе и для физико — химических исследований.
Выполнение работы. На газовом хроматографе снимают хроматограммы полученной контрольной смеси. Условия хроматографирования определяются природой анализируемой смеси. На полученных хроматограммах измеряют расстояния удерживания пиков отдельных компонентов смеси и индивидуальных веществ. Рассчитывают их объемы удерживания. Сравнивая компонентов смеси с индивидуальных веществ, идентифицируют пики контрольной смеси.
Для идентифицированных компонентов смеси рассчитывают удельные удерживаемые объемы. Результаты расчета сводят в таблицу.
Расчет характеристик удерживания
 ,мм
,мм
,мин.
,мл
Работа 2. Определение количественного состава
Цель работы: Определить содержание отдельных компонентов контрольной смеси методом внутренней нормализации без учета калибровочных коэффициентов (методом простой нормировки).
Сущность работы: Для определения количественного состава анализируемой смеси используют зависимость между содержанием данного компонента в смеси и размерами соответствующего ему пика на хроматограмме. Чаще всего количественную оценку хроматограмм производят по площади пиков S. Упрощенный метод измерения площади пика состоит в умножении высоты пика на его ширину, измеренную на расстоянии, равном половине высоты.
Метод простой нормировки основан на предположении, что вещества, независимо от их строения, взятые в одинаковом количестве, дают одну и ту же площадь пика. Это приближено выполняется, если вещества химически сходны, а в качестве газа – носителя применяется газ, теплопроводность которого приблизительно на порядок отличается от теплопроводности анализируемых веществ (детектор – катарометр). Такими обычно являются водород и гелий.
Площадь каждого пика рассчитывают путем умножения высоты пика на его ширину, измеренную на полувысоте пика:
Расчет содержания данного компонента в анализируемой смеси проводят по формуле:
 (2.13),
(2.13),
где ωi – массовая доля  -го компонента, %;
-го компонента, %;  – площадь пика -го компонента, мм2;
– площадь пика -го компонента, мм2;  – сумма площадей пиков всех компонентов, мм2.
– сумма площадей пиков всех компонентов, мм2.
Метод простой нормировки не дает точных результатов в случае различной чувствительности детектора по отношению к разделяемым компонентам смеси.
Выполнение работы: На газовом хроматографе снимают 2–3 воспроизводимых хроматограммы контрольной смеси. Рассчитывают площади пиков отдельных компонентов и определяют содержание компонентов в смеси. Результаты оформляют в виде таблицы.
Расчет состава смеси
Содержание компонента, ω, %
Работа 3. Определение критериев разделения
1. Определить степень разделения 2-х компонентов и селективность жидкой фазы на 2-х колонках.
2. Определить эффективность хроматографических колонок (число теоретических тарелок и ВЭТТ) с различными жидкими фазами.
3. Определить критерии разделения и сравнить две хроматографические колонки по эффективности и селективности.
Сущность работы. Для оценки хроматографического разделения компонентов пользуются тремя группами критериев.
1. Первая группа критериев зависит от природы сорбента и сорбата (разделяемых компонентов), от температуры и характеризует качество разделения в зависимости от различия абсорбируемости или растворимости разделяемых веществ. К критериям этой группы относятся степень разделения  и критерий селективности жидкой фазы КС, которые определяется соотношениями:
и критерий селективности жидкой фазы КС, которые определяется соотношениями:
 2.14),
2.14),
 (2.15),
(2.15),
где  , , –объем, время, расстояние удерживания разделяемых компонентов смеси, соответственно;
, , –объем, время, расстояние удерживания разделяемых компонентов смеси, соответственно;  , , – объем, время, расстояние удерживания несорбирующегося компонента смеси.
, , – объем, время, расстояние удерживания несорбирующегося компонента смеси.
🎬 Видео
Практическая работа 15 Ход определения азота и двуокиси углерода, углеводородных примесейСкачать

6.1. Качественный и количественный анализ спиртов методом газо-жидкостной хроматографииСкачать

Методы количественного определения в фармакопейном анализе.Скачать

Лекция №1_2: Методы количественных определений.Скачать

Хроматография. 1 часть. 10 класс.Скачать

Хроматография ч.1Скачать

Объемные отношения газов при химических реакциях. 8 класс.Скачать

КАК ХРОМИРОВАТЬ ПОВЕРХНОСТЬСкачать

Жидкостной хроматограф, определение содержания ароматических соединений.Скачать
