- Метод внутренней нормализации
- Количественный анализ.
- Метод простой нормировки.
- ОФС.1.2.1.2.0001.15 Хроматография
- ОФС.1.2.1.2.0001.15 Хроматография
- ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
- интерпретация хроматографических данных
- РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
- Метод нормирования (метод внутренней нормализации).
- Метод внешнего стандарта.
- Метод внутреннего стандарта.
- Метод стандартных добавок.
- РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
- ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
- Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
- КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
- Тонкослойная и бумажная хроматография
- Жидкостная хроматография: изократическое элюирование
- Жидкостная хроматография: градиентное элюирование
- Газовая хроматография
- Сверхкритическая флюидная хроматография
- 🎬 Видео
Видео:Хроматэк Аналитик 3.1. Создание метода с нормализациейСкачать

Метод внутренней нормализации
1.4 Метод внутренней нормализации
Этот метод осуществляется в виде нескольких вариантов. В методе простой нормализации сумма площадей всех пиков принимается за 100% и концентрация любого компонента пробы рассчитывается как относительная площадь пика:
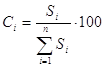
Необходимым условием применения этого метода является регистрация всех компонентов пробы и одинаковая чувствительность детектора к разным веществам. Для большинства детекторов это, в общем, справедливо, если анализируется смесь родственных соединений, молекулярные массы которых значительно не различаются или все компоненты пробы имеют большие молекулярные массы. Например, не требуется калибровка при анализе смеси циклогексана и бензола или при анализе изомеров ксилола. Этот вариант метода имеет ограниченное применение. В большинстве случаев приходится учитывать разный отклик детектора к различным веществам пробы с помощью калибровочных коэффициентов, зависящих от свойств вещества, способа детектирования, а также от конструкции детектора. В основном варианте метода расчет проводится с учетом калибровочных коэффициентов:
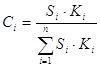
где Сi – концентрация компонента i, %;
Si – площадь соответствующего пика;
Кi – калибровочный коэффициент;
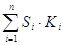 – сумма произведений площадей пиков на относительные поправочные коэффициенты для всех пиков хроматограммы.
– сумма произведений площадей пиков на относительные поправочные коэффициенты для всех пиков хроматограммы.
Достоинства метода внутренней нормализации состоят в том, что он не требует воспроизводимого ввода пробы по величине и тождественности условий анализа. Расчеты проводятся с использованием относительных калибровочных коэффициентов, которые малочувствительны к небольшим изменениям в условиях проведения эксперимента. Недостатки метода заключаются в трудности разделения всех компонентов сложных смесей, необходимости их идентификации и трудоемкости определения калибровочных коэффициентов, хотя некоторые компоненты могут и не представлять аналитического интереса. Недостатком метода является взаимозависимость точности определения одного компонента от точности определения остальных присутствующих в смеси. Ошибки в определении параметров пика или калибровочного коэффициента какого-либо одного компонента приводит к неверным результатам для всех компонентов пробы. Метод используется в основном для рутинных анализов мало-компонентных смесей и для приближенных расчетов.
Недостатки метода заключаются в трудности разделения всех компонентов сложных смесей, необходимости их идентификации и трудоемкости определения калибровочных коэффициентов, хотя некоторые компоненты могут и не представлять аналитического интереса. Недостатком метода является взаимозависимость точности определения одного компонента от точности определения остальных присутствующих в смеси. Ошибки в определении параметров пика или калибровочного коэффициента какого-либо одного компонента приводит к неверным результатам для всех компонентов пробы.
Метод используется в основном для рутинных анализов мало-компонентных смесей и для приближенных расчетов.
2. Техника безопасности и охрана труда
Общие требования безопасности
1.1.1 К самостоятельной работе в лаборатории допускаются лица не моложе 18 лет, имеющие образование не ниже среднего, прошедшие медицинское освидетельствование, вводный инструктаж, первичный инструктаж на рабочем месте, обучение безопасным методам работы и аттестацию по охране труда и промышленной безопасности на допуск к самостоятельной работе.
1.1.2 Работники лаборатории должны соблюдать действующие «Правила внутреннего распорядка для работников ООО «Сибметахим» и «Инструкцию по пропускному и внутри объектовому режиму на охраняемых объектах ООО «Сибметахим».
1.1.3 Режим работы 4-х сменный, выход на работу согласно утвержденному годовому графику учета рабочего времени, для дневных лаборантов режим работы согласно графику учета рабочего времени для дневного персонала.
1.1.4 Для определения состояния здоровья работники лаборатории обязаны проходить 1 раз в год медицинские осмотры.
1.1.5 За работу во вредных условиях труда работникам лаборатории выдается молоко (производство метанола) и талоны спецпитания (производство формалина и карбамидных смол).
1.1.6 Работники лаборатории должны знать:
– свойства имеющихся в лаборатории химических реактивов, продуктов реакции, а также веществ поступающих в лабораторию на анализ, особенно их токсичность, пожароопасность и взрывоопасность;
– опасные моменты при проведении работ в лаборатории и способы их предупреждения;
– профессиональные вредности данной работы и методы борьбы с ними;
– меры первой (доврачебной) помощи при отравлениях, ожогах, поражениях электрическим током, травмах и других несчастных случаях;
– меры по пожарной безопасности и применение первичных
1.1.7 Запрещается выполнение в лаборатории работ, не связанных с заданием и не предусмотренных рабочими инструкциями.
1.1.8 Приточно-вытяжная вентиляция в помещении лаборатории включается за 30 минут до начала работы. При этом в начале включают вытяжную вентиляцию, а потом приточную, а выключают в обратном порядке. Работы в лаборатории должны проводиться только при исправной вентиляции. В случае обнаружения каких-либо неисправностей вентиляции необходимо сообщить об этом начальнику производственной лаборатории химического анализа и контроля за качеством продукции (далее – начальнику лаборатории) или старшему инженеру – химику.
1.1.9 Все работы с газоопасными, летучими или дымящимися веществами следует проводить только в вытяжных шкафах при включенной вентиляции. Шторки вытяжных шкафов следует открывать на минимально удобную для работы высоту, но не более чем на 1/3.
1.1.10 Вытяжные шкафы должны быть оборудованы верхними и нижними отсосами.
1.1.11 Рабочие столы и вытяжные шкафы, предназначенные для работы с пожароопасными и взрывоопасными веществами, должны быть покрыты несгораемыми материалами, а при работе с кислотами, щелочами и другими химически активными веществами – материалами, стойкими к их воздействию, и иметь бортики.
1.1.12 Вытяжные шкафы должны иметь электроосвещение во взрывозащищенном исполнении, электрические выключатели и розетки размещают вне вытяжного шкафа.
1.1.13 При работе в вечернее и ночное время, а также при выполнении особо опасных работ в любое время суток, в лаборатории должно находиться не менее 2-х человек, один из них назначается старшим.
1.1.14 Недопустима работа на неисправном оборудовании. Обо всех неисправностях оборудования, приспособлений, инструмента работник лаборатории должен сообщить начальнику лаборатории или старшему инженеру-химику.
1.1.15 По окончании той или иной операции (работы) необходимо выключить воду, газ, сжатый воздух, электроприборы, применявшиеся при выполнении данной операции.
1.1.16 Запрещается содержать в беспорядке свое рабочее место.
1.1.17 В лаборатории на видном и легкодоступном месте должна находиться аптечка, содержащая необходимые медикаменты для оказания первой помощи и список с их наименованием и назначением.
1.1.18 В случае получения работником лаборатории травмы уведомление администрации и последующие действия выполняются в соответствии с инструкцией ОТБ-11 «О порядке расследования и учета несчастных случаев, микротравм, происшедших с работниками предприятия». Оказание первой помощи в соответствии с инструкцией ОТБ-6 «По оказанию первой помощи пострадавшим».
Видео:Хроматография. Часть 1.Скачать

Количественный анализ.
Практически все методы количественного хроматографического анализа основаны на прямой пропорциональной зависимости параметров пиков от массы вещества в хроматографической зоне. В качестве определяющих параметров используют высоту хроматографического пика (рис. 3.17), его площадь, произведение высоты ника на расстояние удерживания или на время (объем) удерживания. Площадь пика можно рассчитать по следующей формуле:

А для частично разделенных пиков: 
Можно использовать также произведение расстояния (времени и объема) удерживания на высоту пика.

В современных хроматографах хроматограмма выписыва- егся на экране монитора. Время удерживания и площадь пиков измеряются автоматически и выводятся на экран.
Видео:Хроматография. Основы методаСкачать

Метод простой нормировки.
Этот метод основан на предположении, что чувствительность детектора ко всем компоненгам пробы одинакова, т.е. вещества, независимо от их строения, взятые в одинаковом количестве, дают одну и ту же площадь пика. Это приближенно выполняется, если вещества химически сходны, а в качестве газа-носителя применяют газ, теплопроводность которого приблизительно на порядок отличается от теплопроводности анализируемых веществ (дегекгор- катарометр, см. табл. 3.4). Такими газами являются водород и гелий. На полученной хроматограмме исследуемой смеси измеряют высоту каждого пика и ширину его на половине высоты или время удерживания (рис. 3.17). Затем рассчитывают площадь пика или произведение высоты пика на расстояние удерживания. Расчет содержания /-того компонента (со;, %) в анализируемой смеси проводят но формулам:

Метод простой нормировки не дает точных результатов в случае различной чувствительности выбранного детектора но отношению к разделяемым компонентам смеси. Площадь пика на хроматограмме зависит не только от количества вещества в хроматографической зоне, но и определяется характеристиками детектора и условиями проведения анализа. Так, для различных веществ при равной их концентрации в анализируемой смеси на хроматограмме иногда получаются пики неодинаковой площади. Поэтому для проведения количественного анализа недостаточно определить площади хроматографических пиков, а необходимо еще установить для каждого вещества пробы коэффициент пропорциональности между площадью пика и его концентрацией в анализируемой смеси, другими словами, следует провести калибровку детектора в выбранных условиях анализа. Обычно применяют следующие методы калибровки.
Метод нормировки с калибровочными коэффициентами (метод внутренней нормировки). Метод основан на учете различной чувствительности детектора по отношению к комноненгам разделяемой смеси и поэтому более надежен, чем предыдущий. Расчет ведегся но формуле:

где К, — поправочные (градуировочные) коэффициенты, учитывающие чувствительность детектора к данному компоненту.
Если отдельные компоненты разделяются не полностью, что затрудняет измерение площадей, то пользуются другой формулой:

Отдельные поправочные коэффициенты К) определяют следующим образом: готовят модельную смесь соединений, содержащихся в анализируемой смеси (состав ее должен быть близок к составу анализируемой смеси); хроматографируют ее и измеряют площади соответствующих пиков (или произведение времени удерживания на высоту пиков); один из пиков принимают за стандартный и рассчитывают коэффициент но формуле:

где /?г„ тст — масса введенного в хроматограф /-того компонента и стандарта, г;
площадь /-того компонента и стандарта.
Поправочные коэффициенты могут быть как массовыми, так и мольными, в зависимости от способа выражения состава исследуемой смеси, тогда в формулу (3.24) вместо иг, и тап подставляют со,, — воспроизводимого по величине ввода пробы, результаты измерения мало зависят от нестабильности работы хроматографа и детектора, так как эти факторы в равной мере влияют на определяемое и стандартное вещества. Количественный анализ по методу внутреннего стандарта проводят расчетным или графическим способом.
В расчетном способе устанавливают относительный поправочный коэффициент, затем к определенной массе анализируемой пробы добавляют точно измеренное количество внутреннего стандарта. Приготовленную таким образом смесь хроматографируют и измеряют площади /-того компонента и внутреннего стандарта. Расчет проводят но формуле:

где S,, Scm — площади пиков компонента и стандарта; тст, т,ч, — масса стандарта и пробы (без стандарта); кi — относительный поправочный коэффициент.

где К, и Кст — абсолютные градуировочные коэффициенты для компонента и стандарта (см. метод абсолютной градуировки, с. 73).

Рис. 3.23. Градуировочный [рафик в методе внутреннего стандарта
В ?рафическом способе готовят ряд смесей (стандартов) с возрастающим известным массовым или объемным содержанием определяемого компонента; хро- маго1рафируют смеси, измеряют площади компонента и стандарта и строят график зависимости в координатах SCr / Sx — тСТ / mx (рис 3.23). Затем в анализируемую пробу вводят известное количество стандарта, смесь хроматографируют и по графику определяют содержание компонента.
Метод упрощается, если для этой цели в качестве внутреннего стандарта использовать сами определяемые соединения. Такой вариант количественного хроматографического анализа известен под названием метода добавок. Для этого снимают хроматограмму исследуемой смеси и хроматограмму исследуемой смеси с введенным в нее известным количеством определяемого компонента. Измеряют площадь (или высоту) пика анализируемого компонента и рассчитывают содержание вещества по формуле:

При анализе единичных проб можно воспользоваться методом внешнего стандарта, который не требует определения градуировочных коэффициентов и поэтому занимает меньше времени. Записывают хроматограмму исследуемой смеси и одного стандарта с известной концентрацией. После измерения площади ников концентрацию вычисляют по формуле:

Метод дает удовлетворительные результаты, если концентрации стандартного и исследуемого образца сопоставимы, или стандарт имеет хорошее разрешение с ником анализируемого образца и зависимость 5СТ от его концентрации линейна и проходит через начало координат.
Видео:Курс Interlab "Хроматография: толкование и приложения в науке и технологии" Лекция 1Скачать

ОФС.1.2.1.2.0001.15 Хроматография
Содержимое (Table of Contents)
Видео:Хроматография. Часть 3.Скачать

ОФС.1.2.1.2.0001.15 Хроматография
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения.
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
ОБЩАЯ ФАРМАКОПЕЙНАЯ СТАТЬЯ
Хроматография ОФС.1.2.1.2.0001.15
Взамен ст. ГФ XI, вып.1
Хроматографией называется метод разделения смесей веществ, основанный на их многократном перераспределении между двумя контактирующими фазами, одна из которых неподвижна, а другая имеет постоянное направление движения. По механизму, лежащему в основе разделения, различают адсорбционную, распределительную, ионообменную и другие виды хроматографии.
В настоящее время используются следующие хроматографические методы анализа, представленные на рис.1

Рисунок 1- Методы хроматографического анализа.
Результат хроматографического разделения представляется в виде хроматограммы.
Видео:Основы планарной хроматографии: хроматография на бумаге, тонкослойная хроматография (ТСХ), ВЭТСХСкачать

ХРОМАТОГРАММА И ХРОМАТОГРАФИЧЕСКИЕ ПАРАМЕТРЫ
Хроматограмма представляет собой графическое или иное представление сигнала детектора, концентрации веществ в элюате или другой количественной величины, используемой для измерения концентрации веществ в элюате, от времени или объема подвижной фазы. В планарной (плоскостной) хроматографии хроматограммой называют также зафиксированную на бумаге (бумажная хроматография) или ТСХ-пластинке (тонкослойная хроматография) последовательность зон адсорбции веществ исходной (анализируемой) смеси.
Схематически хроматограммы представляют собой последовательность гауссовых пиков на базовой линии (рис. 2).
Базовая линия – сигнал от подвижной фазы.
Пик – часть хроматограммы, регистрирующая отклик детектора. Пик отображает постепенное нарастание концентрации вещества и последующее ее уменьшение. В случае линейной изотермы сорбции кривая, описывающая пик, приближается к кривой гауссова распределения.
Основание пика — продолжение базовой линии, соединяющее начало и конец пика.
Площадь пика (S) – площадь хроматограммы, заключенная между кривой, описывающей пик, и его основанием.
Высота пика (H) – расстояние от максимума пика до его основания, измеренное параллельно оси отклика детектора.

Рисунок 2 — Хроматограмма и основные хроматографические параметры: 1 и 2 – пики соединений 1 и 2; t1 и t2 – соответствующие времена удерживания; t0 – время удерживания несорбирующегося вещества; W1 и W2 – ширина пиков у основания; W0,5 – ширина пика на половине его высоты (предполагается гауссова форма пиков)
Видео:Хроматография ч.1Скачать

интерпретация хроматографических данных
Время удерживания (tR или t) – время, необходимое для элюирования вещества. Соответствует времени появления максимума пика на хроматограмме.
Объем удерживания (VR) – объем подвижной фазы, необходимый для элюирования вещества. Может быть вычислен по времени удерживания и скорости потока (F).

Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока.
Время удерживания несорбирующегося вещества (t0 или tм) – время, необходимое для элюирования неудерживаемого на сорбенте вещества. В эксклюзионной хроматографии t0 соответствует времени удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Объем удерживания несорбирующегося вещества (V0) – объем подвижной фазы, необходимый для элюирования неудерживаемого вещества. Может быть вычислен по времени удерживания неудерживаемого вещества и скорости потока (F):

В эксклюзионной хроматографии V0 соответствует объему удерживания веществ, размер молекул которых больше, чем наибольшие поры сорбента.
Общее время удерживания подвижной фазы (tt) – в эксклюзионной хроматографии время удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Общий объем удерживания подвижной фазы (Vt) – в эксклюзионной хроматографии объем удерживания веществ, молекулы которых меньше, чем наименьшие поры сорбента.
Константа (коэффициент) распределения (K0) – в эксклюзионной хроматографии характеристика элюирования вещества из определенной колонки, которую рассчитывают с помощью выражения:

Приведенное (исправленное) время удерживания вещества (t´R) ‒ время удерживания вещества за вычетом времени удерживания несорбируемого вещества. Может быть рассчитано по формуле:

Исправленное время удерживания не зависит от объема трубопроводов хроматографической системы, установленных между инжектором и колонкой.
Относительное время удерживания (r) – относительное приведенное (исправленное) время удерживания вещества 2 по веществу 1:

Нескорректированное относительное время удерживания (RRT) – относительное время удерживания вещества 2 по веществу 1:

Если не указано иное, значения относительного времени удерживания, приведенные в фармакопейных статьях, соответствуют нескорректированному относительному времени удерживания
Коэффициент емкости (k´) – коэффициент емкости колонки по веществу с временем удерживания tR, показывающий, во сколько раз исправленное время удерживания вещества больше, чем время удерживания несорбируемого вещества.

Эффективность хроматографической системы ‒ параметр, характеризующий степень размывания хроматографического пика. Эффективность выражается числом теоретических тарелок (N):


W – ширина пика у основания;
W0,5 – ширина пика на половине высоты.
При расчете числа теоретических тарелок значения времени удерживания и ширины пика должны быть приведены к одинаковой размерности.
Число теоретических тарелок зависит от природы определяемого вещества, его концентрации или объема, вводимого в систему, от колонки, температуры колонки и состава подвижной фазы.
Если не указано иное, то эффективность хроматографической системы, требования к которой приведено в фармакопейной статье, рассчитывается по формуле, использующей ширину пика на половине его высоты.
Фактор асимметрии (фактор симметрии) пика (As) рассчитывают по формуле:

W0,05 – ширина пика на 5 % (1/20) его высоты;
f – расстояние между перпендикуляром, опущенным из вершины пика, и восходящей стороной пика на 5 % его высоты (рис. 3).
Если фактор асимметрии равен 1, то пик симметричен. Если фактор асимметрии больше 1, то это означает, что растянут задний фронт пика. Если фактор асимметрии меньше 1, то пик растянут спереди.

Рисунок 3 — Схема расчета фактора асимметрии пика
Разрешение (Rs).
Разрешение между пиками двух веществ смеси элюирующимися друг за другом рассчитывают по формулам:


при этом tR2 ≥ tR1. При расчете разрешения величины времени удерживания и ширины пиков должны быть приведены к одинаковой размерности.
Если не указано иное, то разрешение, требования к которому приведены в фармакопейной статье, рассчитывается по формуле, использующей ширину пиков на половине высоты.
В случае, если пики несимметричны и если интенсивность пиков значительно различается, параметр Rs не всегда корректно описывает разделение хроматографических пиков. Таким образом, даже при значениях Rs ≥ 1,5 может наблюдаться неполное разделение пиков. В этих случаях при оценке разделяющей способности можно заменить параметр Rs на параметр «отношение максимум/минимум».
Отношение максимум/минимум (p/v), называемое также отношением «peak-to-valley», «пик – долина». Этот параметр позволяет оценить разделительную способность хроматографической системы. Значение p/v рассчитывается по формуле:

Hp – высота меньшего пика относительно экстраполированной базовой линии;
Hv – высота низшей точки (седловины) кривой, разделяющей пики, относительно экстраполированной базовой линии (рис. 4).

Рисунок 4 — Хроматограмма не полностью разделяемых веществ
Данное соотношение применяется для оценки разделительной способности хроматографической системы, если вещества смеси разделяются не полностью. Рассчитанное отношение максимум/минимум в значительной степени зависит от выбранного варианта интегрирования хроматограммы. Результаты измерения соотношения p/v будут некорректны в случае разметки меньшего пика методом экстраполяции смещенной базовой линии (методом тангенциальной касательной).
Отношение сигнал/шум (S/N).
Краткосрочный шум сигнала детектора (шум базовой линии) влияет на прецизионность количественного определения. Отношение сигнал/шум рассчитывают по формуле:

Н – высота пика, соответствующего рассматриваемому веществу на хроматограмме указываемого стандартного раствора, измеренная от максимума пика до экстраполированной базовой линии. Экстраполяция базовой линии проводится для сигнала на участке базовой линии во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте;
h – размах фонового шума, измеряемый либо на хроматограмме контрольного (холостого) раствора (или раствора плацебо), либо на хроматограмме того же раствора стандартного образца.
Измерение размаха фонового шума проводится во временном интервале, продолжительность которого не менее 5-кратного значения ширины пика на его полувысоте, расположенном, если это возможно, равномерно по обе стороны от места возможного обнаружения пика. В случае использования для измерения шума хроматограммы контрольного (холостого) раствора (или раствора плацебо) измерение размаха фонового шума проводится во временном интервале, включающем в себя время удерживания рассматриваемого вещества, при этом продолжительность временного интервала, в котором проводится измерение шума, должна не менее чем в 5 раз превышать ширину на половине высоты для пика на хроматограмме указываемого раствора стандартного образца (рис. 5).

Рисунок 5 — Вычисление отношения сигнал/шум.
Объем задержки (D) (объем задержки градиента) представляет собой объем системы между точкой, в которой происходит смешение элюентов, и началом колонки.
Объем задержки градиента влияет на времена удерживания веществ и общий профиль наблюдаемой хроматографической картины, получаемой при использовании градиентного элюирования. Объем задержки хроматографической системы определяется следующим способом.
Колонка. Заменяют хроматографическую колонку капилляром (например, 1 м × 0,12 мм).
Подвижная фаза.
Подвижная фаза A — вода.
Подвижная фаза B — 0,1 % (об/об) раствор ацетона.
| Время, мин | Подвижная фаза A, % (об/об) | Подвижная фаза B, % (об/об) |
| 0 – 20 | 100 → 0 | 0 → 100 |
| 20 – 30 | 0 | 100 |
Скорость потока. Необходимая для создания давления, достаточного для стабильной работы насоса (например, 2 мл/мин).
Детектор. Спектрофотометрический при длине волны 265 нм.
Определяют время (t0,5) в минутах, когда оптическая плотность увеличилась на 50 %. Вычисляют объем задержки:
tG – предварительно установленное время градиента (20 мин);
F –скорость потока, мл/мин.

Рисунок 6 — Определение объема задержки градиента
Прецизионность системы в условиях повторяемости
Прецизионность системы в условиях повторяемости выражается в виде рассчитанного относительного стандартного отклонения в процентах [RSD (%)] по результатам последовательных измерений не менее чем 3 вколов или нанесений раствора сравнения и рассчитывается по формуле:

– среднее значение результатов определений;
– результаты единичных определений;
n – число определений.
В планарной хроматографии аналогом времени удерживания является фактор удерживания (Rf):

где a – расстояние от точки нанесения пробы до центра пятна, характеризующего зону адсорбции;
b – расстояние от линии старта до линии фронта элюента.
На экспериментально определяемые значения Rf заметно влияют условия хроматографирования. Оценкой хроматографической подвижности, менее чувствительной к влиянию отклонений в условиях проведения эксперимента, является величина Rst, представляющая собой отношение величины Rf одного вещества к величине Rf другого, принятого за стандарт:

Величины Rf и Rst используют для идентификации веществ. Обычно выбор стандарта осуществляется так, чтобы значение Rst анализируемого вещества было в пределах от 0,5 до 2,0. Схема определения этих величин приведена на рис. 7. 
Рисунок 7 — Схема определения значений Rf и Rst.
 – место нанесения образца на линию старта; 1 – анализируемое вещество (а); 2 – вещество-стандарт (ст); b – расстояние от линии старта до линии фронта элюента.
– место нанесения образца на линию старта; 1 – анализируемое вещество (а); 2 – вещество-стандарт (ст); b – расстояние от линии старта до линии фронта элюента.
Данные планарной хроматографии могут быть представлены в виде денситограмм.
Видео:15x4 - 15 минут про хроматографиюСкачать

РАСЧЕТ СОДЕРЖАНИЯ ОПРЕДЕЛЯЕМЫХ ВЕЩЕСТВ
При расчетах содержания определяемых веществ пики растворителей и реактивов, подвижной фазы или среды (матрицы) образца не учитываются.
Существуют 4 основных метода расчета концентрации анализируемого вещества по хроматографическим данным.
Метод нормирования (метод внутренней нормализации).
Применение данного метода основано на предположении, что на хроматограмме зарегистрированы все вещества, входящие в состав анализируемой смеси, и что доля площади (высоты) каждого пика от суммы площадей (высот) всех пиков соответствует содержанию вещества в массовых процентах. Процентное содержание вещества в анализируемой смеси рассчитывается путём определения площади соответствующего пика как процентной части общей площади всех пиков, за исключением пиков, соответствующих растворителям или реактивам, подвижной фазе или матрице образца. Содержание каждого вещества в смеси в процентах может быть вычислено по формуле:

 – сумма площадей (высот) всех пиков на хроматограмме.
– сумма площадей (высот) всех пиков на хроматограмме.
Если чувствительность детектора различна по отношению к каждому из веществ, то вводят поправочные коэффициенты ki. Относительный коэффициент отклика детектора, обычно называемый фактором отклика, обозначает чувствительность детектора для данного вещества относительно стандартного вещества. Поправочный коэффициент – это число, обратное фактору отклика.
Поправочные коэффициенты рассчитывают относительно основного вещества анализируемой смеси или другого стандартного вещества по формуле:

Сi и С0 – концентрация i-го вещества и стандартного вещества соответственно;
Si и S0 – площадь (высота) пика i-го вещества и стандартного вещества соответственно.
Данные коэффициенты могут не учитываться в случае, если они находятся в пределах диапазона 0,8 – 1,2.
При использовании поправочных коэффициентов выражение для расчета количественного содержания приобретает вид:

При проведении испытания на примеси методом нормализации или методом внешнего стандарта с использованием разведения раствора испытуемого образца в качестве раствора сравнения учитывают все указанные в нормативной документации поправочные коэффициенты, значение которых выходит за пределы диапазона 0,8 – 1,2.
Метод внешнего стандарта.
Концентрацию испытуемого вещества определяют путём сравнения сигнала (пика), полученного на хроматограммах испытуемого раствора, и сигнала (пика), полученного на хроматограммах раствора стандартного образца.
Концентрацию определяемого вещества в испытуемом растворе рассчитывают по формуле:

S и S0 – средние значения площадей (высот) пиков на хроматограммах испытуемого и стандартного растворов соответственно;
С и С0 – концентрации определяемого и стандартного растворов соответственно.
Количественное определение содержания примесей методом внешнего стандарта предпочтительнее проводить с использованием стандартных растворов примесей с концентрациями, близкими к их ожидаемым концентрациям в испытуемом растворе.
В качестве раствора стандартного образца для количественного определения примесей возможно использование раствора основного вещества. В этом случае разведение подбирается таким образом, чтобы концентрация основного соединения в растворе стандартного образца по отношению к его концентрации в испытуемом растворе была близка к ожидаемой концентрации примесей в испытуемом растворе. В этом случае следует учесть факторы отклика примесей по отношению к основному веществу, если их значения выходят за рамки 0,8 — 1,2.
Частным случаем метода внешнего стандарта является метод калибровочной кривой, в ходе которого определяют взаимосвязь между измеренным или обработанным сигналом (у) и количеством (концентрацией, массой и т.д.) определяемого вещества (х) и рассчитывают уравнение калибровочной функции. Результаты испытания рассчитывают из измеренного или обработанного сигнала с помощью обратной функции.
Метод внутреннего стандарта.
Концентрацию определяемого вещества определяют путём сравнения отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме испытуемого раствора и отношения сигналов (площадей или высот пиков), соответствующих определяемому веществу и внутреннему стандарту, на хроматограмме раствора стандартного образца. Метод внутреннего стандарта основан на введении в анализируемую смесь определенного количества стандартного вещества (внутренний стандарт). В испытуемый и стандартный растворы вводят известные количества внутреннего стандарта, хроматографируют растворы и определяют отношения площадей (высот) пиков определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом и стандартном растворах.
Концентрацию определяемого вещества (Х) рассчитывают по формуле:

где  – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в испытуемом растворе;
 – отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в растворе стандартного образца;
– отношение площади (высоты) пика определяемого вещества к площади (высоте) пика внутреннего стандарта в растворе стандартного образца;
С0 – концентрация определяемого вещества в растворе стандартного образца.
В качестве внутреннего стандарта выбирается вещество, отсутствующее в испытуемой пробе, не взаимодействующее с определяемым веществом и другими веществами пробы, обладающее достаточной стабильностью, полностью отделяющееся от веществ пробы и не содержащее примесей с временами удерживания, совпадающими с временем удерживания определяемого вещества. Концентрация внутреннего стандарта должна быть близка к концентрации определяемых веществ, а структура и свойства по возможности аналогичны структуре и свойствам определяемых веществ.
Метод стандартных добавок.
Концентрация определяемого вещества определяется путём сравнения сигнала (площади или высоты пика), соответствующего определяемому веществу, на хроматограмме испытуемого раствора, и сигнала (площади или высоты пика) определяемого вещества на хроматограмме испытуемого раствора с известной добавкой определяемого вещества. Метод стандартных добавок основан на введении в анализируемую смесь известного количества определяемого вещества и сравнения сигналов, полученных для испытуемого раствора со стандартной добавкой и без добавки определяемого вещества. При внесении стандартной добавки стараются минимизировать разбавление испытуемого образца, чтобы измерения раствора со стандартной добавкой и без проходили в одинаковых условиях с одинаковым влиянием матрицы. Количество вводимого в стандартной добавке определяемого вещества должно быть соизмеримо с его предполагаемым содержанием в испытуемом образце. После проведения испытания сравнивают полученные значения интенсивности и рассчитывают количественное содержание определяемого вещества Сх по формуле:

Сстд — концентрация стандартной добавки;
Sx — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора без стандартной добавки;
Sстд+х — интенсивность сигнала определяемого вещества (площадь или высота пика) для испытуемого раствора со стандартной добавкой.
При необходимости формулу корректируют с учетом разбавлений испытуемого раствора за счет введения стандартной добавки.
Видео:Методы очистки и разделения белкаСкачать

РЕКОМЕНДАЦИИ ПО РАЗМЕТКЕ И ИНТЕГРИРОВАНИЮ ХРОМАТОГРАММ ПРИ ОПРЕДЕЛЕНИИ ПРИМЕСЕЙ
Если не указано иное, в испытаниях на содержание примесей в качестве порога игнорирования (неучитываемый предел, уровень содержания вещества, при котором и при меньшем содержании пик не принимается во внимание) принимают величину 0,05 % от содержания основного вещества.
В случаях, когда при испытании на примеси требуется определение суммарного содержания примесей или количественного определения примеси, при интерпретации хроматограммы важно выбрать подходящие условия интегрирования и значения порога интегрирования [интенсивность сигнала пика соединения (высоты или площади)], при котором (и при меньшем) пик не учитывается системой сбора данных и не участвует в количественных расчетах.
С целью разметки на хроматограмме всех пиков, подлежащих учету, заданный порог интегрирования системы сбора данных не должен превышать половину порога игнорирования.
Интегрирование площади пика любой примеси, пик которой не полностью разделяется с основным пиком, предпочтительно проводить экстраполяцией смещенной базовой линии (методом тангенциальной касательной). Использование других способов интегрирования должно быть специально оговорено в фармакопейной статье.
Видео:Опыты по химии. Смеси. Разделение смеси с помощью хроматографииСкачать

ОЦЕНКА ПРИГОДНОСТИ ХРОМАТОГРАФИЧЕСКОЙ СИСТЕМЫ
Испытания пригодности системы являются неотъемлемой частью методики и используются для того, чтобы убедиться в надлежащем функционировании хроматографической системы и обеспечить выполнение предъявляемых к ней требований.
При проведении испытаний используемое оборудование должно быть квалифицировано и способно к функционированию надлежащим образом.
Для оценки пригодности системы указывают:
- требования к параметрам, характеризующим форму пика [эффективность хроматографической системы N и фактор асимметрии (фактор симметрии) As];
- требования к разделительной способности (разрешение между пиками RS или отношение пик — долина p/v);
- требования к воспроизводимости (относительное стандартное отклонение, RSD) значений площади или высоты пиков, а также времен удерживания пиков в случае оценки подлинности соединений;
- требования к чувствительности при проведении испытания на примеси [минимально определяемая концентрация (фактический предел количественного определения) примеси]. Оценивается посредством расчета отношения сигнал/шум для раствора соответствующей концентрации.
Если в фармакопейной статье не указано иное, должны выполняться следующие требования и все дополнительные требования, приведенные в нормативном документе:
- в испытаниях на примеси или количественное содержание для пика на хроматограмме растворе стандартного образца, используемого для количественных определений, значение величины фактора асимметрии (фактора симметрии) As должно находиться в пределах от 0,8 до 1,5;
- разрешение между пиками Rs ³ 1,5 (в случае не полностью разделенных пиков вместо разрешения может быть использовано соотношение p/v. При этом требования к минимальному значению соотношения p/v устанавливаются в фармакопейной статье);
- отношение сигнал/шум для пика вещества, полученное для раствора с концентрацией, равной требуемому уровню минимально определяемой концентрации, должно быть не менее 10. Требуемый уровень минимально определяемой концентрации [фактический предел количественного определения (ПКО)] зависит от того, предполагает ли методика вычисление содержания примесей, или только полуколичественную оценку, когда результат представляется в виде «менее Х» или «не более Х», где Х – допустимое содержание примеси. Для методик, предполагающих вычисление содержания примесей, минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать значение порога игнорирования (если не указано иное – 0,05 % относительно концентрации основного вещества в испытуемом растворе). Для полуколичественных методик минимальная определяемая концентрация для применяемой хроматографической системы не должна превышать максимально допустимое содержание примеси. При необходимости оценки содержания нескольких примесей требуемый уровень минимальной определяемой концентрация для применяемой хроматографической системы определяется примесью, нормы содержания которой наиболее строги. Если в фармакопейной статье не указано иное, то раствор вещества минимально определяемой концентрации для оценки чувствительности детектирования можно приготовить растворением стандартного образца вещества в том же растворителе, который используется для приготовления испытуемого раствора, с уровнем концентрации 0,05 % относительно концентрации основного вещества в испытуемом растворе.
Требования к прецизионности системы в условиях повторяемости при количественном определении основного вещества в субстанциях
При количественном определении основного вещества в фармацевтических субстанциях при содержании основного вещества, близком к 100 %, максимально допустимое относительное стандартное отклонение значений интенсивности (площади или высоты) пика основного вещества для заданного количества повторных введений стандартного раствора RSDmax вычисляется по формуле:

где К – константа (0,349), полученная из выражения:

в котором множитель  соответствует значению RSDmax после 6 повторных инжекций для В = 1,0;
соответствует значению RSDmax после 6 повторных инжекций для В = 1,0;
В – верхний предел содержания основного вещества, приведенный в фармакопейной статье, минус 100 %;
n – число повторных введений стандартного раствора, (3 ≤ n ≤ 6);
t90 %, n‑1 – коэффициент Стьюдента (двусторонний) при доверительной вероятности 90 % с (n – 1) степенью свободы.
Если в фармакопейной статье не указано иное, то значение RSD не должно превышать величин RSDmax, приведенных в таблице. Данное требование не применяется при испытаниях на примеси.
Таблица – Максимально допустимое относительное стандартное отклонение RSDmax в зависимости от верхнего предела содержания основного вещества и числа отдельных введений проб
| В, % | Число отдельных введений проб (вколов) | |||
| 3 | 4 | 5 | 6 | |
| Максимально допустимое относительное стандартное отклонение RSDmax | ||||
| 2,0 | 0,41 | 0,59 | 0,73 | 0,85 |
| 2,5 | 0,52 | 0,74 | 0,92 | 1,06 |
| 3,0 | 0,62 | 0,89 | 1,10 | 1,27 |
Соответствие критериям пригодности системы должно поддерживаться на протяжении всего испытания. В зависимости от различных факторов, например, частоты использования методики и опыта работы с хроматографической системой, аналитик выбирает соответствующую схему проверки, подтверждающую соответствие этим требованиям.
В планарной хроматографии при проведении испытаний, регламентирующих наличие посторонних примесей/родственных соединений, должно быть предусмотрено определение предела обнаружения определяемых примесей и разделительной способности хроматографической системы.
Для подтверждения пригодности системы возможно одновременное использование двух тестов: «Подтверждение разделительной способности» и «Подтверждение чувствительности».
Тест «Подтверждение разделительной способности» проводят путём нанесения на пластинку и последующего хроматографирования специального стандартного раствора, содержащего два или более веществ с известными значениями Rf. После хроматографирования эти вещества должны разделиться, образуя зоны адсорбции, значения Rf которых равны заданным.
Тест «Подтверждение чувствительности» позволяет оценить чувствительность проявления. Он заключается в нанесении определённого количества стандартного вещества, хроматографируемого в условиях предыдущего теста и проявляемого тем же реагентом. Зона адсорбции стандартного вещества должно чётко обнаруживаться.
В каждом случае природа стандартных веществ, состав стандартного раствора, наносимые количества, приблизительные значения Rf устанавливают в фармакопейных статьях.
При необходимости для достижения требуемых критериев пригодности хроматографической системы проводится корректировка хроматографических условий.
Видео:[Запись 2022 г] Введение в хроматографию, основы газовой хроматографииСкачать
![[Запись 2022 г] Введение в хроматографию, основы газовой хроматографии](https://i.ytimg.com/vi/XYD8K73BNyg/0.jpg)
КОРРЕКТИРОВКА УСЛОВИЙ ХРОМАТОГРАФИРОВАНИЯ
Далее приводятся пределы возможных изменений различных параметров хроматографической методики, которые могут быть внесены в нее для соответствия требованиям пригодности системы без принципиального изменения методик. Корректировка условий градиентного элюирования является более критичной, чем изократических условий, так как может вызвать сдвиг пиков на другую стадию градиента, и, таким образом, привести к некорректной идентификации пиков, наложению пиков или сдвигов, при которых элюирование интересующих соединений происходит после указанного времени регистрации хроматограммы. При внесении изменений, отличных от приведенных ниже, требуется проведение повторной валидации методики.
Проверка пригодности системы должна быть включена в методики для подтверждения получения разделения, требуемого для надлежащего проведения испытания. Тем не менее, поскольку неподвижные фазы описаны только в общем виде, и существует огромное количество доступных коммерческих фаз, отличающихся по хроматографическому поведению, некоторая корректировка условий хроматографирования может потребоваться для выполнения требований пригодности системы. В методиках с использованием обращенно-фазовой жидкостной хроматографии корректировки различных параметров не всегда приводят к удовлетворительному разделению. В этом случае может возникнуть необходимость замены одной колонки на другую, такого же типа, обеспечивающую желаемое хроматографическое поведение.
Если в фармакопейной статье не указано иное, допустимы следующие изменения параметров хроматографической системы.
Тонкослойная и бумажная хроматография
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
Пример 1: для вещества, содержание которого составляет 10 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 7 – 13 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению абсолютного содержания до 8 – 12 %. Допустимое изменение по относительному содержанию больше, чем допустимое изменение по абсолютному содержанию. Таким образом, допускается изменение содержания 7 – 13%.
Пример 2: для вещества, содержание которого составляет 5 % подвижной фазы, изменение относительного содержания на 30 % приведет к изменению его абсолютного содержания до 3,5 – 6,5 %, тогда как изменение на 2 % по абсолютному содержанию приведет к изменению его абсолютного содержания в подвижной фазе до 3 – 7 %. В этом примере допустимое изменение по абсолютному содержанию больше, чем допустимое изменение по относительному содержанию, и допустимым диапазоном содержания является 3 – 7 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Наносимый объём. При использовании пластинок с малым размером частиц сорбента (2 — 10 мкм) наносимый объём может быть изменен на 10 –20 % от заявленного объёма.
Жидкостная хроматография: изократическое элюирование
Состав подвижной фазы: количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше. Абсолютное содержание других веществ не может быть изменено более чем на 10 %.
pH среды водного компонента подвижной фазы: ± 0,2 pH, если иное не указано в нормативном документе, или ± 1,0 pH в случаях испытания неионизируемых веществ.
Концентрация солей в буферном веществе подвижной фазы: ± 10 %.
Скорость потока подвижной фазы: ± 50 %, большая степень изменений допустима при одновременном изменении размеров колонки. При необходимости одновременного изменения размеров колонки и скорости потока сначала рассчитывается номинальная скорость потока для колонки, размеры которой отличаются от приведенной в нормативном документе, а затем допускается корректировка полученного значения скорости потока на ± 50 %.
- замена типа неподвижной фазы недопустима (например, недопустима замена фазы С18 на фазу С8);
- размер частиц: максимальное уменьшение размера частиц 50 %, увеличение не допускается.
Длина колонки: ± 70 %,
внутренний диаметр колонки: ± 25 %.
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

F1 — скорость потока, указанная в нормативном документе, мл/мин;
F2 — скорректированная скорость потока, мл/мин;
l1 — длина колонки, указанная в нормативном документе, мм;
d1 — внутренний диаметр колонки, указанный в нормативном документе, мм;
d2 — внутренний диаметр используемой колонки, мм.
Температура: ± 10 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы (минимально определяемая концентрация) и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Жидкостная хроматография: градиентное элюирование
Изменение хроматографических условий для систем с градиентом требует большей осторожности, чем для изократических.
Состав подвижной фазы/профиль градиентного элюирования: незначительные изменения состава подвижной фазы и системы градиента являются приемлемыми при условии, что:
— выполнены требования пригодности системы;
— основной пик элюируется в пределах ± 15 % от обозначенного времени удерживания;
— элюирующая способность конечного состава подвижной фазы не менее предписанного состава.
Если при использовании градиентного элюирования не может быть достигнуто соответствие требованиям пригодности системы, рекомендуется оценить объем задержки градиента или сменить используемую колонку.
Объем задержки градиента. Конфигурация используемого оборудования может значительно изменить разрешение, указанные абсолютные и относительные времена удерживания. Это может произойти из-за отличия объема задержки градиента используемой системы от объема задержки градиента системы, с помощью которой проводилась валидация методики. В нормативном документе часто перед началом программы градиента включена изократическая стадия, чтобы можно было адаптировать систему к временным точкам градиента с учетом разницы объема задержки у системы, использованной для разработки методики, и у фактически использующейся системы. Решение о необходимости адаптации длительности изократической стадии для данного аналитического оборудования принимается пользователем. Если объем задержки, использованный при разработке методики, приведен в нормативном документе, то интервалы времени (t), указанные в таблице градиента, можно заменить адаптированными интервалами времени (tc), рассчитанными с помощью следующего уравнения:

где D – объем задержки, мл;
D0 – объем задержки, использованный при разработке методики, мл;
F – скорость потока, мл/мин.
Изократическая стадия, введенная с этой целью, может быть исключена, если имеются данные валидации по применению методики без этой стадии.
pH среды водного компонента подвижной фазы. Изменения не допускаются.
Концентрация солей в буферном веществе подвижной фазы. Изменения не допускаются.
Скорость потока. Изменения допустимы при изменении размеров колонки.
— замена типа неподвижной фазы недопустима (например, недопустима замена С18 на С8);
— размер частиц: изменения не допускаются;
— длина колонки: ± 70 %;
— внутренний диаметр колонки: ± 25 %;
При изменении размеров колонки скорость потока пересчитывают, используя следующее уравнение:

где F1 — скорость потока, указанная в нормативном документе, мл/мин;
F2 — скорректированная скорость потока, мл/мин;
l1 — длина колонки, указанная в нормативном документе, мм;
d1— внутренний диаметр колонки, указанный в нормативном документе мм;
d2 — внутренний диаметр используемой колонки, мм.
Температура. ± 5 % от указанной рабочей температуры, если не указано иначе.
Длина волны детектора. Изменения не допускаются.
Вводимый объём пробы. Может быть уменьшен при условии, что детектирование (предел количественного определения) и сходимость отклика (RSD площадей или высот) для пика (пиков) определяемых соединений остаются удовлетворительными.
Газовая хроматография
— размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки);
— толщина плёнки: от минус 50 % до плюс 100 % (капиллярные колонки).
-Длина колонки: ± 70 %;
-Внутренний диаметр колонки: ± 50 %.
Скорость потока: ± 50 %.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
Сверхкритическая флюидная хроматография
Состав подвижной фазы. Для набивных колонок количество растворителя, содержание которого в смеси наименьшее, может изменяться в пределах ± 30 % (относительное содержание) или ± 2 % (абсолютное содержание) в зависимости от того, какая из величин будет больше; для систем с капиллярной колонкой изменения не допускаются.
Длина волны детектора. Изменения не допускаются.
— размер частиц: максимальное уменьшение на 50 %, увеличение не допускается (набивные колонки).
— Длина колонки: ± 70 %;
— Внутренний диаметр колонки: ± 25 % (набивные колонки), ± 50 % (капиллярные колонки).
Скорость потока: ± 50 %.
Температура: ± 5 %, если температура указана в нормативном документе.
Вводимый объём пробы. Может быть уменьшен при условии, что чувствительность фактически применяемой хроматографической системы и прецизионность системы в условиях повторяемости для определяемых соединений остаются удовлетворительными.
🎬 Видео
Хроматография за 5 минут | БИОЛОГИЯ ЕГЭ | СОТКАСкачать

6.1. Качественный и количественный анализ спиртов методом газо-жидкостной хроматографииСкачать

Хроматография. 1 часть. 10 класс.Скачать

Высокоэффективная жидкостная хроматографияСкачать

Жидкостной хроматограф, определение содержания ароматических соединений.Скачать

Лабораторная работа по ионообменной хроматографииСкачать

Хроматин. Типы хроматина. Упаковка генетического материала. Эухроматин, гетерохроматинСкачать

Вебинар: "Возможности и примеры использования ГХ/МС в лабораторной диагностике"Скачать
