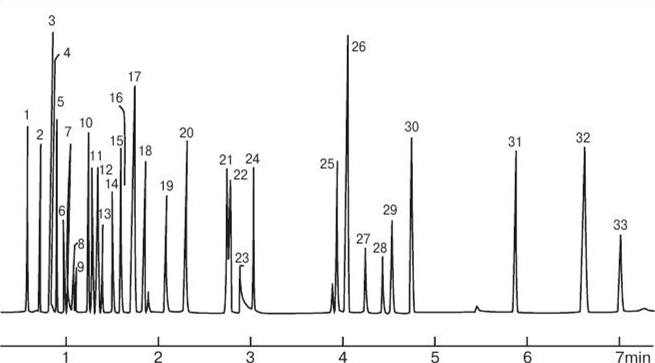
- Параметры хроматографических пиков
- Главные параметры пиков
- Типы хроматографических пиков
- Автоматическая разметка пиков
- Ширина
- Пороги
- Ручная разметка пиков
- Идентификация пиков
- Идентификация по абсолютному времени удерживания
- Идентификация по относительному времени удерживания
- Идентификация по относительному объему удерживания
- Идентификация по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах
- Идентификация по индексам удерживания (Ковача)
- Идентификация по температурам кипения
- Основные Параметры Хроматографических Пиков
- Что такое хроматографический пик
- Автоматическая и ручная разметка пиков
- Как идентифицировать пики
- Заключение
- Хроматографические методы анализа. Учебно-методическое пособие (стр. 3 )
- Характеристики удерживания
- 🎥 Видео
Видео:Хроматография. Часть 1.Скачать

Параметры хроматографических пиков
Процесс обработки хроматографического сигнала состоит из нескольких этапов: фильтрация шумов, разметка пиков, идентификация пиков, градуировка, количественный расчет. В этой статье мы рассмотрим один из наиболее важных вопросов — разметку пиков и их основные параметры. Мы расскажем как происходит разметка пиков на примере программы NetChrom, разработанной компанией «Мета-хром».
- Главные параметры пиков
- Типы хроматографических пиков
- Автоматическая разметка пиков
- Ручная разметка пиков
- Идентификация пиков:
- по абсолютному времени удерживания
- по относительному времени удерживания
- по относительному объему удерживания
- по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах
- по индексам удерживания (Ковача)
- по температурам кипения
Видео:Хроматэк Аналитик 3.1. Создание метода с градуировкойСкачать

Главные параметры пиков
Разметка пиков (интегрирование) — операция вычисления параметров пиков, полученных на хроматограмме путем определения характерных точек (начало, вершина и конец пика). При этом пики ограничиваются базовой линией (прямой, соединяющей точки начала и конца пика на нулевой линии), которая проводится по методу «резиновой ленты», натягивающейся снизу от точки начала до точки конца базовой линии на протяжении всей хроматограммы.
На основании вычислений оцениваются основные параметры пика:
- время удерживания — время от начала анализа до максимума пика;
- площадь — область, заключенная между пиком и ограничивающей его базовой линией;
- ысота — расстояние между базовой линией и максимумом пика;
- ширина пика на половине его высоты.
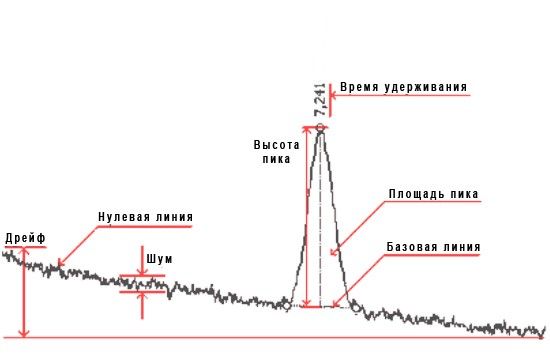
Важно! Особенностью программы NetChrom является полностью автоматизированный процесс ее настройки под конкретный хроматографический сигнал, непрерывное отслеживание изменения характеристик хроматографического сигнала в ходе обработки хроматограммы: уровня шумов и дрейфа, а также уровня самого сигнала.
Для определения характерных точек пиков: начала, конца и вершины — в программе NetChrom используется алгоритм на основе вычисления первой сглаженной производной хроматографического сигнала, скорректированной на дрейф базовой линии.
При превышении или достижении определенного уровня этой производной, называемой порогом, определяются характерные точки пиков. Уровень порога определяется на основе шума производной на участках хроматографического сигнала, свободного от пиков.
Кроме уровня шума, дрейфа и уровня сигнала, большое значение имеет также зависимость изменения ширины хроматографических пиков от времени их удерживания в процессе анализа (ширина пика в начале и в конце анализа).
Как известно, ширина хроматографических пиков непрерывно увеличивается в процессе анализа, причем в изотермическом режиме эта зависимость имеет линейный характер, в режиме программирования температуры колонок эта зависимость имеет более сложный характер. Программа использует эту зависимость для автоматической настройки программных фильтров под хроматографический сигнал во время анализа.
Кроме этого, пользователю предоставлена возможность сформировать или отредактировать зависимость изменения ширины хроматографических пиков от времени удерживания самостоятельно (ширина пиков в начале и в конце хроматограммы).
При неправильном задании этой зависимости возможно некорректное определение характерных точек пика и даже пропуск небольших пиков. Зависимость изменения ширины хроматографических пиков от времени индивидуальна для каждого метода, и после изменения условий анализа: температуры колонок, расхода газоносителя и др., необходимо откорректировать зависимость.
Видео:Пирогов А.В. Газовая хроматографияСкачать
Типы хроматографических пиков
Пики могут быть нескольких типов:
- простые пики — начало и конец пика принадлежат базовой линии;
- хвостатые пики — асимметрия заднего фронта пика;
- пики наездники — пики на хвосте большего по величине пика;
- неразделенные пики — конец первого пика совпадает с началом второго и эта точка не принадлежит базовой линии;
- зашкаленные пики — пики с плоской вершиной.
Разделение слившихся пиков производится по перпендикуляру или тангенте в зависимости от соотношения ширины и высоты этих пиков.
Пики наездники отделяются по тангенте и площадь под ними включается в площадь хвостатого пика.
Следует иметь в виду, что никакой алгоритм не может в ряде случаев гарантировать корректную разметку на пики, поскольку само понятие «пик» во многом субъективно и зависит от конкретной аналитической задачи. Например, нельзя гарантировать корректную разметку на пики при сложной форме базовой линии, плохом разделении хроматографических пиков, малых пиках-наездниках, высоком уровне шумов и так далее.
При этом правильность получаемых результатов зачастую зависит от опыта пользователя. В программе NetChrom реализованы два подхода для проведения разметки пиков:
- автоматическая разметка пиков;
- ручная (графическая) разметка пиков.
Ниже мы рассмотрим каждый из подходов более детально.
Видео:Введение в спектрофотометриюСкачать
Автоматическая разметка пиков
Автоматическая разметка пиков (интегрирование) имеет смысл тогда, когда ожидается обработка серии хроматограмм со сходными, повторяющимися особенностями базовой линии, значениями величины и последовательностью хроматографических пиков.
Параметры интегрирования, с помощью которых пользователь может влиять на процесс обнаружения пиков на хроматограмме, представлены ниже:
Ширина

Пороги
Порог обнаружения пиков может быть задан по двум параметрам: минимальной площади и минимальной высоте пика.
Минимально допустимая площадь детектируемого пика. При детектировании пиков имеется возможность не размечать или подавлять пики, площадь которых меньше заданной. При этом значение параметра, равное 0, означает, что подавление пиков выключено.

- Минимальная высота
Минимально допустимая высота детектируемого пика. Подавление пиков с высотой значение которой, меньше заданного. При этом значение параметра, равное 0, означает, что подавление пиков выключено.

Не всегда получается подобрать одинаковые параметры для разметки всех пиков. Например, с увеличением времени выхода пика, изменяется его ширина, поэтому значения параметров, подходящие для пиков в начале хроматограммы, могут не подходить для пиков в ее конце. В таких случаях рекомендуется использовать события интегрирования.
Настройка алгоритма разметки с использованием событий интегрирования имеет смысл, если ожидается серия однотипных хроматограмм со сходными, повторяющимися особенностями базовой линии. События интегрирования позволяют настроить процесс разметки в соответствии с особенностями данной серии хроматограмм, задавая для некоторых участков хроматограмм индивидуальные параметры и правила разметки.
Событие можно задать для отдельного детектора, и оно начинает действовать с указанного момента времени до тех пор, пока не будет переопределено другим событием такого же типа, или пока не завершится хроматограмма. Если не удается добиться желаемой разметки при использовании параметров и событий интегрирования, используют ручное графическое интегрирование (непосредственно на графике хроматограммы).
Видео:Ошибки при построении градуировкиСкачать

Ручная разметка пиков
Ручная разметка пиков используется, если не удается добиться желаемой разметки при использовании параметров автоматической разметки. Ручная графическая разметка пиков производится непосредственно на графике хроматограммы. При этом все действия, выполняемые пользователем, относятся к выделенному фрагменту хроматограммы или к выделенному пику. В программе существуют следующие типовые операции по ручному редактированию пиков на хроматограмме:
- создание пика;
- разделение пика;
- слияние пиков;
- корректировка положения характерных точек пика;
- задание пика наездником;
- задание пика хвостатым;
- задание пика базовым;
- задание пика слившимся;
- удаление пика;
- удаление пиков справа;
- удаление пиков слева;
- удаление всех пиков.
Видео:[Запись 2022 г] Введение в хроматографию, основы газовой хроматографииСкачать
![[Запись 2022 г] Введение в хроматографию, основы газовой хроматографии](https://i.ytimg.com/vi/XYD8K73BNyg/0.jpg)
Идентификация пиков
Идентификация — отнесение пиков на хроматограмме к тому или иному компоненту из списка компонентов рабочего метода. При этом производится сравнение рассчитанных параметров удерживания всех обнаруженных на хроматограмме пиков с информацией, хранящейся в таблице компонентов. Идентификация компонентов по одному или по нескольким детекторам осуществляется следующими способами:
- по абсолютному времени удерживания;
- по относительному времени удерживания;
- по относительному объему удерживания;
- по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах;
- по индексам удерживания (Ковача);
- по температурам кипения.
Теперь рассмотрим каждый из вышеперечисленных способов подробнее.
Идентификация по абсолютному времени удерживания
Наиболее простой способ идентификации — по времени удерживания, то есть сравнение времени удерживания анализируемого компонента со временем удерживания известного соединения при строго заданных условиях анализа. Для проведения идентификации пика по времени удерживания в библиотеке компонентов должна содержаться информация:
- наименование компонента;
- время удерживания;
- окно поиска по времени (в единицах времени).
Окно поиска — границы области, в которой будет осуществляться поиск пика как в положительную, так и в отрицательную сторону от заданного в таблице параметра удерживания.
При задании окна необходимо стремиться, чтобы его ширина была достаточной для попадания пика в окно при неизбежных изменениях времени удерживания, но и не слишком большой, чтобы в него не попадали соседние пики.
В случае если в окно поиска попадают несколько пиков, то среди них выбирается пик, имеющий максимальную вероятность идентификации (наиболее интенсивный или ближайший к библиотечному времени).
Идентификация по относительному времени удерживания
При изменении условий в процессе анализа (расход газа-носителя, температура колонки), а также в процессе «старения» колонок, пики могут не попасть в окно поиска. Это происходит чаще всего:
- когда нельзя задать достаточно широкое окно поиска, чтобы в него не попали соседние пики;
- при длительных анализах с программированием температуры колонок, когда время удерживания может измениться в большей степени.
Выход из положения состоит в том, что один из пиков (или несколько) назначается «стандартом времени», для него задается увеличенное окно поиска (2-5%), а для остальных пиков рассчитывается относительное время удерживания.
Стандартом времени, как правило, выбираются стоящие отдельно или большие пики обязательно присутствующие на хроматограмме. Таким образом, при одновременном сдвиге по той или иной причине времен удерживания всех компонентов, наличие пиков-стандартов времени поможет правильно идентифицировать вещества, несмотря на то, что их время удерживания не будет попадать в окно поиска по времени.
В этом случае идентификация производится следующим образом:
- производится поиск пика стандарта времени по времени удерживания;
- для стандарта времени удерживания рассчитывается коэффициент отклонения реального времени удерживания по сравнению с библиотечным временем по формуле:
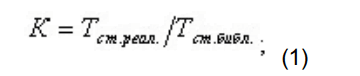
для остальных пиков рассчитывается ожидаемое время удерживания, исходя из времени, заданного в библиотеке для данного компонента, и рассчитанного коэффициента отклонения времени стандарта по формуле:
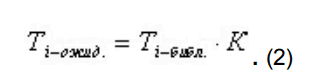
Идентификация по относительному объему удерживания
Является более точным расчетным параметром по сравнению с относительным временем удерживания, так как в нем учитывается время на прохождение подвижной фазой расстояния от устройства для ввода пробы до детектора (иногда это время называют «мертвым» временем или временем удерживания несорбирующегося вещества).
При этом относительный объем удерживания стандарта времени принимают за единицу, а относительный объем удерживания компонентов (Ri) рассчитывают по формуле:
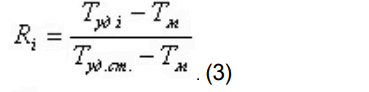
T уд i — время удерживания анализируемого компонента;
T уд.ст. — время удерживания стандарта времени;
T м — мертвое время.
Идентификация по времени удерживания и соотношению интенсивностей пиков на параллельно (последовательно) работающих детекторах
Если в процессе анализа используются несколько (чаще всего два) параллельно или последовательно работающих детектора, то для более достоверной идентификации можно применить способ идентификации, заключающийся в том, что наряду со временем удерживания (абсолютным или относительным), можно использовать отношение интенсивностей пиков соответствующих детекторов.
Вначале пик идентифицируется по времени удерживания на ведущем детекторе (детекторе, по интенсивности пика которого рассчитывается концентрация компонента). Затем сравниваются отношения интенсивностей пика на различных детекторах с библиотечным отношением.
Для идентификации может использоваться не только отношение интенсивностей, но также наличие или отсутствие пика на другом (не ведущем) детекторе.
Идентификация по индексам удерживания (Ковача)
Для идентификации могут использоваться и другие относительные параметры удерживания, которые в меньшей степени зависят (в отличие от времени удерживания) от условий анализа. Одним из таких параметров является индекс удерживания — безразмерная величина, характеризующая положение пика вещества на хроматограмме относительно пиков выбранных стандартов.
Если в качестве стандартов используются насыщенные углеводороды (алканы, парафины), то индекс удерживания называется индексом Ковача. Выбор типа индекса (линейный или логарифмический) зависит от условий анализа.
Для постоянной температуры колонки во время анализа характерна логарифмическая зависимость, при программировании — линейная. Однако между этими двумя зависимостями нет четкого разделения, поэтому при применении режима программирования температуры колонок с начальным изотермическим участком, используется смешанный тип индекса. При этом на изотермическом участке выбирается логарифмический тип индекса, до первого реперного пика, который попадает на участок программирования температур, и в дальнейшем — линейный тип индекса.
Начало программирования температуры колонок выбирается согласно заданным параметрам управления соответствующего метода. При идентификации по индексам удерживания в таблицу компонентов должны быть занесены табличные значения индекса компонентов и ширина окна поиска по индексу. Пикам – стандартам индексов удерживания необходимо присвоить тип: реперный в таблице компонентов и задать увеличенное окно поиска (2-5%) от времени удерживания.
Идентификация по индексам удерживания производится следующим образом:
- Производится идентификация реперных пиков по времени удерживания.
- Реперным пикам присваиваются соответствующие индексы из таблицы компонентов.
- Используя индексы удерживания реперных пиков, рассчитываются индексы удерживания обычных пиков и сравниваются с табличными данными.
Индексы удерживания Ковача рассчитываются по формулам:
линейный индекс удерживания:
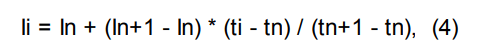
логарифмический индекс удерживания:
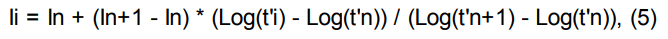
Ii — время удерживания интересующего пика;
In, In+1 — индексы предыдущего и последующего компонентов с известной величиной индекса;
ti — время удерживания интересующего пика;
tn, tn+1 — времена удерживания пиков, соответствующие предыдущему и последующему компонентам с известными индексами;
t’. — приведенное время удерживания.
Важно! Для увеличения точности расчета индексов удерживания в качестве времени удерживания необходимо брать приведенное время удерживания. Приведенное время удержания равно разности абсолютного времени удержания и мертвого времени (времени нахождения неудерживаемого компонента в хроматографической системе).
Мертвое время определяется либо экспериментально, либо расчетом, выполненным с помощью газового калькулятора. В качестве неудерживаемого компонента чаще всего используют метан.
В окно ожидаемого времени или индекса удерживания одного компонента может попасть несколько пиков. В этом случае выбор пика, в зависимости от настройки, может осуществляться по следующим критериям:
- ближайший к ожидаемому (библиотечному) времени или индексу удерживания;
- максимальный по высоте;
- максимальный по площади.
Один и тот же пик может попасть в окна поиска разных компонентов, в этом случае необходимо уменьшить ширину окон поиска.
Указанная схема распознавания (идентификации) компонентов оказывается достаточно универсальной и гибкой для того, чтобы проблема корректного распознавания пиков в подавляющем большинстве случаев было решена. Все, что требуется от оператора — на этапе настройки грамотно выбрать пик стандарта времени или реперные пики и в дальнейшем периодически корректировать их ожидаемые времена удерживания по текущей хроматограмме.
Идентификация по температурам кипения
Частным случаем расчета индексов удерживания является расчет температур кипения. При расчете температур кипения используются те же формулы, что и при расчете индексов. Для расчета температур кипения необходимо в библиотеке методов выбрать тип индекса (линейный или логарифмический) и задать известные температуры кипения реперных компонентов.
Видео:Хроматография ч.1Скачать

Основные Параметры Хроматографических Пиков
 21.01.2021
21.01.2021
Ключевую для хроматографии информацию получают при анализе пиков на полученной диаграмме. Хроматограммы бывают двух видов: полученные при планарной хроматографии или при жидкостной. Во втором случае, как правило, анализ проводится специальным детектором, анализирующем проходящую через колонку смесь.
Видео:Хроматэк Кристалл: Детектор ПФДСкачать

Что такое хроматографический пик
Хроматографический пик представляет собой резкое возрастание концентрации вещества с последующим уменьшением до базовой линии. Происходит это в момент прохождения вещества через детектор. Пики тем четче, чем больше разделительная способность системы и чем сильнее различаются свойства компонентов смеси, и в теории представляют собой гауссовские кривые.
Различные вещества имеют различные пики ввиду индивидуальных свойств. Вследствие неоднородной реакции с подвижной фазой время прохождения через колонку компонентов разнится. Каждый пик на диаграмме отражает отдельное вещество или группу веществ, прошедших колонку за определенное время.
Время прохождения вещества зависит от его сродства к подвижной и неподвижной фазе. Само же сродство определяется протекающими между веществом и фазами реакциями, размером компонента смеси, температурой и скоростью элюента. Поэтому, для каждой системы абсолютное время прохождения веществ разное, и зависит от каждого параметра анализа.
После установки основных характеристик пиков — время удерживания, высота и площадь пика – переходят к их соотношению. Отношение площадей пиков равно отношению количеств веществ компонентов смеси.
Высота пика в случае с гауссовскими кривыми также может отражать относительное содержание вещества в пробе. Это применимо для неперекрывающихся симметричных пиками. В противном случае расчеты, основанные на высоте пика, ведут к ошибкам.
Ширина пика показывает эффективность хроматографической системы, ведь чем больше ширина пика, тем хуже полученные данные о его площади. Чем меньше в смеси примесей и чем больше степень разложения на компоненты, тем эффективнее анализ. Ширина также увеличивается в процессе анализа. При постоянной температуре это происходит линейно.
На практике полученные диаграммы содержат отклонения от теоретических расчетов. Причина этому реакция среды, наличие примесей, изменение температуры во время анализа, схожие свойства самих веществ. Пики могут накладываться друг на друга, быть ассиметричной формы. Чем дольше идёт анализ, тем шире они становятся. Расчеты, основывающиеся на высоте асимметричных пиков, ведут к ошибкам, ведь они больше не отражают реальной концентрации.
Пики отделяются от шумов, которые возникают при детальном рассмотрении базовой линии. Как и любое другое электрическое устройство, хроматограф имеет отклонения показаний в одну и другую сторону. Чем меньше выбранный промежуток времени, тем менее заметны сами шумы.
Видео:Презентация Маэстро-АльфаМС для сотрудников ФГБУН Института химии растворов им. Г. А. Крестова РАНСкачать

Автоматическая и ручная разметка пиков
Ввиду того, что теоретически число пиков неограниченно, необходимо отделять пики от шумов. Делать это можно как вручную, так и при помощи специальных программ. Автоматизация уместна в случае с большим количеством сходных хроматограмм. При этом для каждой серии анализов требуется индивидуальная настройка.
В первую очередь подавляются шумы. Происходит это при помощи введения порогового значения для высоты и площади пиков. Так, зная степень разделения смеси средой, вводят минимальную высоту. А на основании данных об ориентировочном содержании веществ вводится минимальная площадь.
После назначения порогов назначается эталонная ширина пика. От этого параметра зависит, какая часть пика будет интегрироваться. Пики с меньшей шириной также автоматически отсеиваются. Этот процесс облегчает дальнейший анализ и упрощает полученные данные.
Подбор этих значений должен производиться с расчетом на то, что со временем анализа пики расширяются. Имеет смысл деление хроматограммы на участки с индивидуальными параметрами разметки. Подбор специальных значений для порогов и ширины пиков повышает качество полученных данных.
К ручной разметке прибегают в случаях, когда автоматизация не предоставляет искомой точности. Исследователь собственноручно выделяет необходимые пики, однако последующий подсчет площади под ними производится автоматически. Идентифицировать вещества по их пикам же все равно приходится вручную.
Видео:Расчет RRF и анализ образцаСкачать

Как идентифицировать пики
После выделения пиков производится идентификация, соотношение сигналов с веществами, которые их дали. Достичь качественного результата позволяет сравнение со стандартным образцом. Однако доказать наличие конкретного вещества сложнее, чем доказать его отсутствие. Ведь если пик на характерной для определенного вещества отметке отсутствует, то это однозначно говорит об отсутствии самого вещества, а его наличие может свидетельствовать о смеси других веществ с аналогичными свойствами.
Зафиксировав четыре абсолютных значения, производят анализ как на непосредственно полученных данных, так и на относительных величинах. Минусом абсолютных значений является высокая чувствительность к колебаниям таких параметров как температура и скорость подвижной фазы, что приводит к неточностям и необходимости многократных анализов.
Простейшим методом идентификации является соотношение времени удерживания вещества в пробе и в стандарте. Сопоставив полученные и библиотечные времена удерживания, делают вывод о наличии или отсутствии искомых веществ. Для подтверждения точности полученных данных результаты должны быть повторяемы.
Для получения более точных результатов прибегают к измерению относительного объема удерживания. Здесь учитывается время прохождения колонки подвижной фазы. Объем удерживания, в отличие от времени удерживания, не зависит от скорости потока, что и делает этот параметр универсальной характеристикой. Иногда прибегают к комбинированию детекторов. Сравнив пики на нескольких хроматограммах, делают вывод о свойствах вещества. Наличие пиков на одних и отсутствие на других детекторах помогает идентифицировать вещество.
Видео:Курс Interlab "Хроматография: газовая хроматография. Детекторы." Лекция 5Скачать

Заключение
Несмотря на то, что зачастую пики не соответствуют гауссовским кривым, идентификация возможна и в сложнейших случаях. Современные библиотеки располагают количеством информации, которое позволяет точно определять искомые вещества даже в смеси со схожими свойствами компонентов.
Автоматизированная разметка пиков и их интегрирование снимает с работающего за хроматографом нагрузку, облегчая аналитическую задачу и повышая эффективность процесса. Заглушая шумы и считая площади пиков, программное обеспечение снижает общее время анализа.
Проведенный анализ позволяет дать количественную характеристику изучаемой смеси, обнаружить в ней искомые вещества, что делает хроматографию востребованной во многих сферах.
![]()

Генераторы азота и сфера их применения
В газовой хроматографии азот –…
31.01.2022

Газовые хроматографы: устройство и принцип работы.
Любой газовый хроматограф состоит минимум…
30.01.2022
ГХ или ВЭЖХ? Что выбрать?
При появлении новой аналитической задачи…
16.11.2021
Хроматография. Простыми словами.
О хроматографии написано много. Мы…
10.11.2021

Как проводится хроматография
Хроматографический анализ представляет собой один…
18.03.2021

Абсорбционная спектрометрия уже больше века…
18.03.2021
Основные Параметры Хроматографических Пиков
Ключевую для хроматографии информацию получают…
21.01.2021

Результатом хроматографии является хроматограмма, дающая…
21.01.2021

Распространённые причины поломки хроматографов
Использование любых сложных видов оборудования…
02.10.2020

Как Хроматография Применяется в Парфюмерии?
Методику хроматографии активно используют в…
02.10.2020

Хроматография: история открытия и развития
Хроматография сегодня активно используется в…
06.09.2020

Как правильно выбрать хроматограф?
Хроматография – метод анализа жидкостных…
05.09.2020

Работа любого сложного устройства сопровождается…
28.07.2020

Сегодня хроматография остается самым используемым…
28.07.2020

Предшественником всех современных спектрометров считается…
06.07.2020

Разделение сложных смесей на единичные…
06.07.2020

Хроматографические методы в криминалистике
Криминалистические экспертизы играют важную роль…
06.07.2020
Хроматография в фармацевтической промышленности
В настоящее время можно выделить…
27.05.2020

Принципы работы спектрометра
Спектрометр – прибор, работающий на…
08.05.2020

Хромато-масс-спектрометры: принцип действия
Командой Хроматограф.ру в Печорской центральной…
08.05.2020

Порядок технического обслуживания оборудования производства «НПО СПЕКТРОН»
При поставке приборы снабжаются всем…
17.04.2020

Хроматография в контроле качества продовольственного сырья и пищевых продуктов
Безопасность и качество продуктов питания…
17.04.2020
Телемедицина для хроматографов
Что такое телемедицина? Это консультация…
15.04.2020

Основные производители хроматографов в мире, в России
Хроматографы используются в аналитических исследованиях,…
02.12.2019

Области применения газовых и жидкостных хроматографов
Хроматография – способ разделения многокомпонентных…
02.12.2019

Хроматографические Методы Анализа
Хроматографические методы анализа базируются на…
02.12.2019

Хроматограф — принцип действия, виды хроматографов
Одним из самых популярных методов…
23.02.2019
7 (343) 300 90 95 обратный звонок
info@gcpro.ru написать письмо
Екатеринбург, Сибирский
тракт, 57 оф. 308
Видео:Занятие 10. ФотометрияСкачать

Хроматографические методы анализа. Учебно-методическое пособие (стр. 3 )
 | Из за большого объема этот материал размещен на нескольких страницах: 1 2 3 4 5 |

Этот простой и точный метод является основным методом определения микропримесей. Кроме того, метод не требует разделения всех компонентов смеси, а ограничивается лишь теми, определение которых необходимо в данном конкретном случае.
Метод внутреннего стандарта основан на введении в анализируемую смесь точно известного количества стандартного вещества. В качестве стандартного выбирают вещество, близкое по физико-химическим свойствам к компонентам смеси. Это вещество должно отсутствовать в исследуемой смеси и давать на хроматограмме пик, отдельный от других компонентов. После хроматографирования измеряют площади пиков анализируемого компонента (Si) и стандартного вещества (SCT).Массовую долю компонента (Wi, %) рассчитывают по формуле:
 (1.17),
(1.17),
где r – отношение массы внутреннего стандарта к массе пробы.
Достоинством метода внутреннего стандарта является хорошая воспроизводимость, высокая точность, отсутствие влияния на измеряемые величины небольших колебаний условий опыта.
К недостаткам относятся требование точной дозировки стандарта и хорошего отделения пика стандарта от пиков анализируемых веществ. Пользование калибровкой возможно только для той области концентраций, в которой сохраняется линейная зависимость между показаниями детектора и концентрацией определяемого вещества.
Метод простой нормировки чаще всего используют на практике. Для его использования необходимо, чтобы на хроматограмме были зарегистрированы все компоненты, входящие в состав анализируемой смеси; сумму площадей всех пиков принимают за 100 %. Тогда отношение площади одного пика к сумме площадей, умноженное на 100, будет характеризовать массовую долю (%) компонента в смеси.
Этот метод основан на том предположении, что вещества, взятые в одинаковом количестве, дают одну и ту же площадь пика, независимо от их строения. Это приближенно выполняется, если вещества химически сходны, а в качестве газа-носителя применяется газ с высокой теплопроводностью (водород или гелий).
Если чувствительность детектора различна по отношению к разделяемым компонентам смеси, то используют метод нормировки с поправочными коэффициентами. В этом случае расчет ведут по формуле (1.18), где ki – поправочный коэффициент i-го компонента (мг/см2):
 (1.18)
(1.18)
Поправочные коэффициенты получают при анализе стандартных серий и рассчитывают по формуле
 (1.19)
(1.19)
где С – концентрации определяемого и стандартных веществ. Метод нормировки требует полного разделения и идентификации всех компонентов смеси; необходимости в знании калибровочных коэффициентов для всех без исключения компонентов смеси. Ошибки в определении параметра пика или калибровочного коэффициента какого-либо одного компонента приводят к неверным результатам всего анализа. Поэтому метод нормировки применяется главным образом для рутинных анализов малокомпонентных смесей и для приближенных результатов.
5.8. Практическое применение газовой хроматографии
Газовая хроматография – один из наиболее перспективных физико-химических методов анализа. В настоящее время вряд ли существует научно-исследовательская или производственная лаборатория, занимающаяся анализом органических веществ, в которой отсутствовала бы хроматографическая аппаратура.
Методом газовой хроматографии анализируют нефтяные и рудничные газы, воздух, продукцию основной химии и промышленности основного органического синтеза, нефть и продукты ее переработки, металлоорганические соединения и т. д. Газовая хроматография используется в биологии, медицине, в технологии переработки древесины, в лесохимии и пищевой промышленности.
Выпускаемая аппаратура позволяет анализировать не только вещества, представляющие собой в нормальных условиях газы, но и высококипящие соединения, фармацевтические препараты, различные пестициды и т. д.
Газовая хроматография применяется также для автоматизации производственных процессов. Датчик промышленного хроматографа используется не только как регистрирующий прибор, но и как регулирующее устройство, подающее сигналы непосредственно исполнительным механизмам. Таким образом, промышленный хроматограф может контролировать и регулировать важнейшие параметры технологического процесса: температуру, давление, расход сырья и т. д.
Из физико-химических применений газовой хроматографии следует отметить возможность изучения термодинамики сорбции, определения молекулярных масс, давления пара веществ, коэффициентов диффузии, поверхности адсорбентов и катализаторов.
Важной особенностью газовой хроматографии является возможность определения в различных продуктах микропримесей. В настоящее время методом газовой хроматографии удается определять концентрации порядка 10-10%. Это делает метод незаменимым при анализе мономеров, используемых в производстве полимерных материалов, а также при исследовании биосферы.
Вопросы для самоконтроля
1. Что такое: а) общее время удерживания; б) приведенное (исправленное) время удерживания; в) общий удерживаемый объем; г) приведенный (исправленный) удерживаемый объем?
2. В чем сущность качественного хроматографического анализа по величине удерживаемого объема?
3. Почему предпочитают использовать исправленный удерживаемый объем, а не объем удерживания?
4. Какие хроматографические параметры можно использовать для идентификации компонентов смеси?
5. Почему идентификацию компонентов не рекомендуется вести по абсолютным временам удерживания?
6. Какие другие способы идентификации компонентов применяют в хроматографическом анализе?
7. Можно ли сделать вывод о природе веществ на основании хроматографических данных?
8. Как зависит время (объем) удерживания от растворимости соединения в подвижной фазе?
9. Что такое относительный удерживаемый объем и относительное время удерживания?
10. В чем сущность основных методов количественной хроматографии: а) нормировки с поправочными коэффициентами; б) абсолютной калибровки; в) внутреннего стандарта?
11. Укажите возможности и ограничения различных количественных методов хроматографического анализа.
12. Как можно измерить площадь пика на хроматограмме? Какой зависимостью связана площадь пика с концентрацией вещества?
13. Почему способ абсолютной калибровки сравнительно редко применяют в хроматографических лабораториях?
14. Как повысить точность определения компонента по методу нормировки?
15. Какие параметры хроматографичеcкого пика используют для количественного анализа?
16. В каких случаях в количественном хроматографическом анализеизмеряют высоту пика? площадь пика?
6. Жидкостная колоночная хроматография
Жидкостная хроматография (ЖХ) – это метод разделения и анализа сложных смесей веществ, в котором подвижной фазой служит жидкость. Метод ЖХ применим для разделения более широкого круга веществ, чем метод ГХ, поскольку большинство веществ не обладает летучестью, многие из них неустойчивы при высоких температурах. В ЖХ разделение чаще всего происходит при комнатной температуре. Жидкая подвижная фаза, в отличие от газа в ГХ, выполняющего только транспортную функцию, является активным элюентом. Молекулы жидкой фазы могут сорбироваться на поверхности неподвижной фазы. При прохождении через колонку находящиеся в элюенте молекулы интересуюшего нас компонента должны вытеснить молекулы элюента с поверхности сорбента. Применяя различные элюенты, можно изменять параметры удерживания и селективность хроматографической системы.
В классическом варианте ЖХ в стеклянную колонку длиной 1–2 м, заполненную сорбентом (размер частиц  100 мкм), вводят анализируемую пробу и пропускают элюент. Скорость прохождения элюента под действием силы тяжести мала, а продолжительность анализа значительна. Однако такой вариант ЖХ не требует дорогостоящего оборудования и до сих пор находит применение.
100 мкм), вводят анализируемую пробу и пропускают элюент. Скорость прохождения элюента под действием силы тяжести мала, а продолжительность анализа значительна. Однако такой вариант ЖХ не требует дорогостоящего оборудования и до сих пор находит применение.
Вследствие использования сорбентов со значительно меньшим размером частиц (до 5–10 мкм), нагнетательных насосов, чувствительных детекторов произошел переход от классической к высокоэффективной жидкостной хроматографии (ВЭЖХ), позволяющей проводить разделение и определение молекул, ионов, разделение макромолекул и биологически активных молекул. К достоинствам метода ВЭЖХ можно отнести универсальность, возможность автоматизации разделения и анализа сложных смесей органических и неорганических веществ, экспрессность, эффективность и высокую чувствительность. Это серийный метод определения органических соединений многих классов, его широко используют при анализе смесей аминокислот, белков, лекарственных препаратов. ВЭЖХ находит применение и в неорганическом анализе для разделения ионов в зависимости от их размера.
6.1. Адсорбционная хроматография
В адсорбционном варианте жидкостной хроматографии в зависимости от полярности неподвижной и подвижной фаз различают нормально-фазовую (НФХ) и обращенно-фазовую (ОФХ) хроматографии. В НФХ используют полярный адсорбент и неполярные подвижные фазы. В ОФХ – неполярный адсорбент и полярные подвижные фазы. Неподвижная фаза должна удерживать разделяемые компоненты. Подвижная фаза, т. е.растворитель, должна обеспечить различную емкость колонки и эффективное разделение за приемлемое время.
Неподвижные фазы. В качестве адсорбентов применяют тонкодисперсные пористые материалы.
Полярные адсорбенты ( ,
,  , оксиды металлов, флорисил и др.) имеют на поверхности слабокислотные ОН-группы, способные удерживать вещества с основными свойствами. Недостаток полярных сорбентов – высокая чувствительность к содержанию воды в растворителях, приводящая к изменению свойств поверхности и невоспроизводимым результатам анализа. Для ВЭЖХ применяют полярные сорбенты с привитыми полярными группами (амины, диолы и др.), что позволяет менять селективность, подбирая подходящий элюент.
, оксиды металлов, флорисил и др.) имеют на поверхности слабокислотные ОН-группы, способные удерживать вещества с основными свойствами. Недостаток полярных сорбентов – высокая чувствительность к содержанию воды в растворителях, приводящая к изменению свойств поверхности и невоспроизводимым результатам анализа. Для ВЭЖХ применяют полярные сорбенты с привитыми полярными группами (амины, диолы и др.), что позволяет менять селективность, подбирая подходящий элюент.
Неполярные адсорбенты (графитированная сажа, кизельгур, диатомит) не проявляют селективности к полярным молекулам. Используют также сорбенты с привитыми неполярными фазами, например силикагель с алкилсилильными группами от С2 до С22.
Подвижные фазы. В ЖХ важен выбор подвижной фазы, поскольку она оказывает большое влияние на селективность разделения, эффективность колонки и скорость движения хроматографической полосы. Подвижная фаза должна растворять анализируемую пробу, обладать малой вязкостью, из нее должно быть возможным выделение разделенных компонентов. Подвижная фаза должна быть инертна по отношению к материалам всех частей хроматографа, безопасной, дешевой.
Разделение компонентов достигают, меняя элюирующую силу растворителя.
Элюирующая сила растворителя показывает, во сколько раз энергия сорбции данного элюента больше, чем энергия сорбции элюента, выбранного в качестве стандарта, например н-гептана.
Растворители (элюенты) делят на слабые и сильные. Слабые растворители слабо адсорбируются неподвижной фазой, поэтому коэффициенты распределения сорбируемых веществ (сорбата) высокие. Сильные растворители сильно адсорбируются, поэтому коэффициенты распределения сорбата низкие. Растворитель тем сильнее, чем выше растворимость в нем анализируемой пробы, чем сильнее взаимодействие растворитель–сорбат.
Элюирующая сила определяется полярностью растворителя. В НФХ с увеличением полярности растворителя элюирующая сила растворителя растет, в ОФХ – снижается. Часто применяют не индивидуальные растворители, а их смеси. Незначительные добавки другого растворителя, особенно воды, существенно увеличивают элюирующую силу элюента.
При разделении многокомпонентных смесей одна подвижная фаза в качестве элюента может не разделить все компоненты пробы. В этом случае применяют метод ступенчатого или градиентного элюирования, применяя в процессе хроматографирования последовательно все более сильные элюенты. Установлены некоторые эмпирические правила, помогающие при выборе элюента. Сорбция, как правило, увеличивается с ростом числа двойных связей и ОН-групп в соединениях. Сорбция уменьшается в ряду органических соединений: кислоты–спирты–альдегиды–кетоны–сложные эфиры–ненасыщенные углеводороды–насыщенные углеводороды.
Для разделения веществ разной полярности и для разделения соединений разных классов применяют НФХ. В ОФХ неподвижная фаза сильнее адсорбирует неполярные компоненты из полярных элюентов, например из воды.
Метод адсорбционной ВЭЖХ – это серийный метод определения органических соединений многих классов, его широко используют при анализе смесей аминокислот, белков, лекарственных, препаратов.
6.2. Распределительная хроматография.
Метод распределительной, или жидкостно-жидкостной, хроматографии основан на распределении вещества между двумя несмешивающимися жидкостями, подобно тому, как это происходит в многократной ступенчатой экстракции. Жидкую неподвижную фазу наносят на пористый достаточно инертный сорбент и заполняют им распределительную колонку. При пропускании жидкой подвижной фазы через колонку смесь разделяется на компоненты главным образом за счет их различной растворимости в жидкой неподвижной фазе. Обычно растворимость компонентов пробы в подвижной и неподвижной фазах, обладающих разной полярностью, сильно различается. Если растворимость пробы выше в неподвижной фазе, то время удерживания компонентов значительно возрастает. Если растворимость пробы выше в подвижной фазе, то время удерживания может быть близким к времени удерживания несорбируемого компонента. Чтобы добиться разделения, в подвижную фазу, насыщенную неподвижной, включают третий компонент, снижающий различие в полярности подвижной и неподвижной фаз. Например, к смеси из неполярного (гексан) и полярного (вода) растворителей прибавляется спирт.
В нормально-фазовой распределительной хроматографии используют следующие системы: полярный растворитель (вода, спирт) фиксирован на твердом носителе – силикагеле, диатомите, целлюлозе, оксиде алюминия. Полярной фазой в этом случае служат неполярные растворители – изооктан, бензол, и др.
В обращенно-фазовой распределительной хроматографии неполярный растворитель фиксируют на носителе, а в качестве подвижной фазы используют полярные растворители (вода, спирт, буферные растворы, сильные кислоты).
Нанесенные жидкие фазы имеют большой недостаток – они быстро смываются подвижной жидкой фазой с поверхности носителя, особенно, при использовании таких систем в ВЭЖХ, т. е. при повышенном давлении в колонке. Поэтому жидкие фазы прививают к носителю. В качестве носителей неподвижных жидких фаз для НФРХ используют силикагели с привитыми нитрильными, аминными и другими группами. В обращенно-фазовом варианте используют силикагели с привитыми алкилсилильными группами. Механизм удерживания на таких сорбентах сложен.
Метод распределительной хроматографии применяют для разделения сильнополярных соединений, аминокислот, фенолов, фенилкарбоновых кислот и др.
6.3. Эксклюзионная хроматография
Эксклюзионная хроматография – это разновидность жидкостной хроматографии, в которой разделение компонентов основано на распределении молекул в соответствии с их размером между растворителем, находящимся в порах сорбента, и растворителем, протекающим между его частицами. В процессе разделения небольшие молекулы попадают в сетку полимера, в порах которой растворитель служит неподвижной фазой, и удерживаются там, большие молекулы не могут проникнуть в полимерную сетку и вымываются из колонки подвижной фазой. Вначале элюируются самые большие, затем средние и потом небольшие молекулы. Поэтому эксклюзионную хроматографию называют также молекулярно-ситовой. Эксклюзионная хроматография подразделяется на гель-проникающую и гель-фильтрационную. В гель-проникающей хроматографии разделение осуществляется на полимерах, набухающих в органических растворителях; если же полимеры набухают в воде, то говорят о гель-фильтрационном варианте.
Каждый сорбент характеризуется объемом пор, следовательно, областью разделяемых молекулярных масс и градуировочным графиком, который имеет сложный вид, характеризующий зависимость удерживаемого объема от молекулярной массы или размера молекул. Надо подбирать сорбент и длину хроматографической колонки такими, чтобы разделение вещества протекало в пределах линейного участка градуировочного графика.
Неподвижные фазы в эксклюзионной хроматографии выбирают для конкретной аналитической задачи. Первоначально устанавливают, какая система растворителей может быть использована для анализа (водная или водно-органическая), что и определяет тип сорбента.
Подвижные фазы в эксклюзионной хроматографии должны удовлетворять определенным требованиям:
– полное растворение образца;
– хорошее смачивание сорбента;
– низкая вязкость и токсичность.
Метод эксклюзионной хроматографии широко используют при исследовании полимеров, определении их молекулярных масс, а также в биологии и медицине для анализа белков, крови и других объектов.
6.4. Особенности жидкостных хроматографов.
Жидкостной хроматограф – более сложный прибор по сравнению с газовым. Это связано с тем, что система подачи элюента включает ряд дополнительных узлов: систему дегазации, градиентное устройство, насосы и измерители давления.
Градиентное устройство должно обеспечить отбор элюентов из двух-трех емкостей в смеситель, затем в колонку. Насосы должны иметь постоянную скорость потока от 0.1 до 10 мл/мин при давлении 400 атм. Кроме того, необходимо тщательное удаление газа из всех используемых растворителей, так как появление пузырьков газа в детекторе недопустимо. Проба вводится с помощью петлевых дозаторов или специальных микрошприцов через прокладку из специальных ненабухающих полимерных материалов.
В ВЭЖХ обычно используют прямые колонки длиной 10, 15, 25 см с внутренним диаметром 4–5,5 мм. В микроколочных хроматографах используют колонки длиной 5–6 см и диаметром 1–2 мм. Колонки изготавливают из стекла или нержавеющей стали.
В ионном хроматографе все соединительные трубки, колонки, краны выполнены из химически инертных материалов, что позволяет использовать сильнокислотные и сильноосновные элюенты.
Для непрерывного контроля элюата, вытекающего из колонки, обычно используют дифференциальные рефрактометры, люминесцентные, УФ-спектрофотометрические и кондуктометрические детекторы.
Дифференциальный рефрактометр – это универсальный детектор, который позволяет определять общий показатель преломления системы проба– элюент, т. е. сигнал дают все компоненты, показатель преломления которых отличается от показателя преломления элюента. Чувствительность детектора – 10-6 г.
УФ-детектор работает при одной и той же длине волны, соответствующей наиболее интенсивной линии ртутной лампы низкого давления  =253.7 нм. УФ-детектор наиболее чувствителен, если молярные коэффициенты светопоглощения компонентов высоки, а элюент не поглощает в ультрафиолетовой области спектра. С помощью такого детектора можно определять любые ароматические соединения, большинство кетонов и альдегидов. УФ-детектор селективен, чувствительность его составляет 10-9 г.
=253.7 нм. УФ-детектор наиболее чувствителен, если молярные коэффициенты светопоглощения компонентов высоки, а элюент не поглощает в ультрафиолетовой области спектра. С помощью такого детектора можно определять любые ароматические соединения, большинство кетонов и альдегидов. УФ-детектор селективен, чувствительность его составляет 10-9 г.
Фотометры и спектрофотометры позволяют работать при любой длине волны (190-650 нм). Регистрируют изменение поглощения во времени при определенной длине волны или в остановленном потоке элюента снимают спектр. Быстрозаписывающий спектрофотометр позволяет записать всю спектральную область за 20с.
Кондуктометрический детектор применяют в ионной хроматографии для измерения проводимости раствора, пропорциональной числу ионов в растворе, их подвижности. Предел обнаружения с помощью такого детектора составляет порядка 10-3 мкг/мл. Использование концентрирующей колонки позволяет снизить предел обнаружения на 2–3 порядка.
В эксклюзионной хроматографии при анализе полимеров для определения средних молекулярных масс используют проточный нефелометр.
Вопросы для самоконтроля
1. Какова роль подвижной фазы в жидкостной хроматографии?
2. Какими способами проба анализируемой смеси веществ вводится в хроматографическую установку в жидкостной хроматографии?
3. Какова роль основных узлов в жидкостном хроматографе высокого давления? Что общего и каковы принципиальные отличия от газового хроматографа?
4. Назовите три способы детектирования в жидкостной хроматографии.
5. Почему в жидкостной хроматографии предпочитают подвижные фазы с низкой вязкостью?
6. Какие варианты используются в жидкостно-жидкостной распределительной хроматографии?
7. Каковы особенности эксклюзионной хроматографии?
8. Как изменяется время (объем) удерживания молекул в эксклюзионной хроматографии с увеличением их размера?
9. Чем отличаются нормально — и обращенно-фазовый варианты ВЭЖХ?
ГЛАВА 2. ПРАКТИЧЕСКИЕ РАБОТЫ
Работа 1. Определение качественного состава смеси на основе
Цель работы: Идентифицировать компоненты хроматографируемой смеси по объемам удерживания и для идентифицированных компонентов рассчитать удельные удерживаемые объемы.
Cущность работы: Газовая хроматография позволяет проводить индивидуальную и групповую идентификацию веществ (т. е. отнесение их к определенной группе соединений). Индивидуальную хроматографическую идентификацию проводят с помощью следующего приема: сравнивают характеристики удерживания компонентов анализируемой смеси с характеристиками удерживания стандартов, компонентов стандартных смесей или с табличными данными.
Видео:Хроматография. 1 часть. 10 класс.Скачать

Характеристики удерживания
1. Расстояние удерживания  , мм – расстояние от момента ввода пробы на хроматограмме до максимума соответствующего пика.
, мм – расстояние от момента ввода пробы на хроматограмме до максимума соответствующего пика.  – расстояние удерживания несорбирующегося компонента, мм, определяется от момента ввода пробы до максимума пика несорбирующегося компонента;
– расстояние удерживания несорбирующегося компонента, мм, определяется от момента ввода пробы до максимума пика несорбирующегося компонента;
2. Исправленное (приведенное) расстояние удерживания,  – расстояние от вершины пика несорбирующегося компонента до максимума соответствующего пика, мм;
– расстояние от вершины пика несорбирующегося компонента до максимума соответствующего пика, мм;
 (2.1)
(2.1)
3. Время удерживания  ,мин – время, прошедшее от момента ввода пробы в колонку до момента записи максимума соответствующего пика.
,мин – время, прошедшее от момента ввода пробы в колонку до момента записи максимума соответствующего пика.
Время удерживания можно рассчитать, зная линейную скорость движения диаграммной ленты в потенциометре Uл, мм/мин:
 (2.2)
(2.2)
(часто время удерживания измеряется непосредственно с помощью секундомера).

Рис. 2.1. Хроматограмма, иллюстрирующая расстояние удерживания
4. Исправленное время удерживания 
 , мин – время, прошедшее с момента появления максимума пика несорбирующегося компонента до появления максимума пика соответствующего соединения,
, мин – время, прошедшее с момента появления максимума пика несорбирующегося компонента до появления максимума пика соответствующего соединения,
 (2.3),
(2.3),
где  – время удерживания несорбирующегося компонента.
– время удерживания несорбирующегося компонента.
Время удерживания есть функция длины колонки, скорости потока газа-носителя, сорбируемости, температуры. Величиной, не зависящей от скорости потока газа-носителя, является объем удерживания.
5. Объем удерживания (удерживаемый объем),  , мл – объем газа- носителя при температуре колонки, прошедшего через колонку за время от момента ввода пробы до момента регистрации максимума соответствующего пика хроматограммы.
, мл – объем газа- носителя при температуре колонки, прошедшего через колонку за время от момента ввода пробы до момента регистрации максимума соответствующего пика хроматограммы.
 (2.4),
(2.4),
где  –исправленная объемная скорость газа носителя, мл/мин.
–исправленная объемная скорость газа носителя, мл/мин.
Объемная скорость газа-носителя  измеряется на выходе из колонки жидкостным расходомером. Для расчета истинного (исправленного)значения, , следует учитывать давление насыщенного пара рабочей жидкости расходомера (обычно воды),
измеряется на выходе из колонки жидкостным расходомером. Для расчета истинного (исправленного)значения, , следует учитывать давление насыщенного пара рабочей жидкости расходомера (обычно воды),  , при температуре расходомера
, при температуре расходомера  , тогда,
, тогда,
 (2.5)
(2.5)
где — измеренная объемная скорость расхода газа-носителя на выходе из колонки, мл/мин.;  — давление газа на выходе из колонки, обычно 1 атм;
— давление газа на выходе из колонки, обычно 1 атм;  – температура колонки, К.
– температура колонки, К.
6. Приведенный (исправленный) объем удерживания  , мл – объем удерживания с поправкой на объем удерживания несорбирующегося компонента (
, мл – объем удерживания с поправкой на объем удерживания несорбирующегося компонента ( ). Рассчитывается по формуле:
). Рассчитывается по формуле:
 (2.6)
(2.6)
7. Эффективный (истинный) объем удерживания  ,мл – приведенный объем удерживания, исправленный с учетом перепада давления в колонке:
,мл – приведенный объем удерживания, исправленный с учетом перепада давления в колонке:
 (2.7),
(2.7),
где j – поправочный коэффициент, учитывающий сжимаемость газа носителя в колонке:
 (2.8)
(2.8)
где  – давление газа-носителя на входе в колонку, атм;
– давление газа-носителя на входе в колонку, атм;  – давление газа-носителя на выходе из колонки, равное 1 атм.
– давление газа-носителя на выходе из колонки, равное 1 атм.
Таким образом, истинный удерживаемый объем равен:
 (2.9)
(2.9)
6. Удельный объем удерживания при температуре колонки,  – истинный объем удерживания, отнесенный к единице массы неподвижной жидкой фазыв колонке. Рассчитывается по формуле:
– истинный объем удерживания, отнесенный к единице массы неподвижной жидкой фазыв колонке. Рассчитывается по формуле:
 (2.10),
(2.10),
где q – масса неподвижной жидкой фазы в колонке, г.
9. Удельный объем удерживания  – удельный объем удерживания при температуре колонки, приведенный к 273 К, рассчитывается по формуле:
– удельный объем удерживания при температуре колонки, приведенный к 273 К, рассчитывается по формуле:
 (2.11)
(2.11)
В газожидкостной хроматографии удельный удерживаемый объем представляет собой физико-химическую константу. Значение ее зависит только от природы жидкой фазы и не зависит от условий хроматографического разделения. Величина может быть использована для идентификации компонентов в качественном анализе и для физико — химических исследований.
Выполнение работы. На газовом хроматографе снимают хроматограммы полученной контрольной смеси. Условия хроматографирования определяются природой анализируемой смеси. На полученных хроматограммах измеряют расстояния удерживания пиков отдельных компонентов смеси и индивидуальных веществ. Рассчитывают их объемы удерживания. Сравнивая компонентов смеси с индивидуальных веществ, идентифицируют пики контрольной смеси.
Для идентифицированных компонентов смеси рассчитывают удельные удерживаемые объемы. Результаты расчета сводят в таблицу.
Расчет характеристик удерживания
 ,мм
,мм
,мин.
,мл
Работа 2. Определение количественного состава
Цель работы: Определить содержание отдельных компонентов контрольной смеси методом внутренней нормализации без учета калибровочных коэффициентов (методом простой нормировки).
Сущность работы: Для определения количественного состава анализируемой смеси используют зависимость между содержанием данного компонента в смеси и размерами соответствующего ему пика на хроматограмме. Чаще всего количественную оценку хроматограмм производят по площади пиков S. Упрощенный метод измерения площади пика состоит в умножении высоты пика на его ширину, измеренную на расстоянии, равном половине высоты.
Метод простой нормировки основан на предположении, что вещества, независимо от их строения, взятые в одинаковом количестве, дают одну и ту же площадь пика. Это приближено выполняется, если вещества химически сходны, а в качестве газа – носителя применяется газ, теплопроводность которого приблизительно на порядок отличается от теплопроводности анализируемых веществ (детектор – катарометр). Такими обычно являются водород и гелий.
Площадь каждого пика рассчитывают путем умножения высоты пика на его ширину, измеренную на полувысоте пика:
Расчет содержания данного компонента в анализируемой смеси проводят по формуле:
 (2.13),
(2.13),
где ωi – массовая доля  -го компонента, %;
-го компонента, %;  – площадь пика -го компонента, мм2;
– площадь пика -го компонента, мм2;  – сумма площадей пиков всех компонентов, мм2.
– сумма площадей пиков всех компонентов, мм2.
Метод простой нормировки не дает точных результатов в случае различной чувствительности детектора по отношению к разделяемым компонентам смеси.
Выполнение работы: На газовом хроматографе снимают 2–3 воспроизводимых хроматограммы контрольной смеси. Рассчитывают площади пиков отдельных компонентов и определяют содержание компонентов в смеси. Результаты оформляют в виде таблицы.
Расчет состава смеси
Содержание компонента, ω, %
Работа 3. Определение критериев разделения
1. Определить степень разделения 2-х компонентов и селективность жидкой фазы на 2-х колонках.
2. Определить эффективность хроматографических колонок (число теоретических тарелок и ВЭТТ) с различными жидкими фазами.
3. Определить критерии разделения и сравнить две хроматографические колонки по эффективности и селективности.
Сущность работы. Для оценки хроматографического разделения компонентов пользуются тремя группами критериев.
1. Первая группа критериев зависит от природы сорбента и сорбата (разделяемых компонентов), от температуры и характеризует качество разделения в зависимости от различия абсорбируемости или растворимости разделяемых веществ. К критериям этой группы относятся степень разделения  и критерий селективности жидкой фазы КС, которые определяется соотношениями:
и критерий селективности жидкой фазы КС, которые определяется соотношениями:
 2.14),
2.14),
 (2.15),
(2.15),
где  , , –объем, время, расстояние удерживания разделяемых компонентов смеси, соответственно;
, , –объем, время, расстояние удерживания разделяемых компонентов смеси, соответственно;  , , – объем, время, расстояние удерживания несорбирующегося компонента смеси.
, , – объем, время, расстояние удерживания несорбирующегося компонента смеси.
🎥 Видео
КАК ХРОМИРОВАТЬ ПОВЕРХНОСТЬСкачать

6.1. Качественный и количественный анализ спиртов методом газо-жидкостной хроматографииСкачать

Курс Interlab "Хроматография: газовая хроматография. Ввод пробы. Часть 2." Лекция 7Скачать

Вебинар: "Возможности и примеры использования ГХ/МС в лабораторной диагностике"Скачать

Газовая хроматомасс-спектрометрия. Часть 1Скачать

Разделяющая молекулы, объединяющая людей. Что такое хроматографияСкачать
