Видео:Фармакокинетика: всасывание, распределение, биодоступность лекарственных препаратовСкачать

Основные принципы фармакокинетики
Основные фармакокинетические параметры
Концентрация лекарственного вещества (С) — это ее количество в определенном объеме крови в конкретный момент времени после введения в организм.
Объем распределения препарата (Vd — volume of distribution) — характеризует степень захвата лекарственного вещества (ЛВ) тканями из плазмы крови и выражается формулой Vd = D / Co — это условный объем жидкости, который требуется для равномерного распределения введенной дозы ЛВ до концентрации, которая определяется в крови в момент исследования. Различают также понятие удельный объем распределения — это отношение объема распределения к массе тела, выражается в л/кг. Объем распределения ЛВ в известной мере определяет степень проникновения его из крови и внеклеточной жидкости в ткани, а также создание его депо в органах.
Площадь под фармакокинетической кривой «концентрация — время» (AUC — area under the curve) — площадь фигуры, ограниченная фармакологической кривой и осями координат (АUC = Co / Kel). Площадь под кривой определяется объемом распределения и общим клиренсом лекарственного средства. Биодоступность (F) определяют относительным количеством ЛВ, поступающим в системный кровоток и взаимодействующим с тканевыми рецепторами. Биодоступность препарата при внесосудистом введении определяют как соотношение к его лекарственной форме для внутрисосудистого введения. Если исследуют лекарственные формы для внесосудистого и внутривенного введения в равных дозах, применяют формулу:
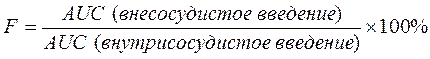
Биоэквивалентность (сравнительная биодоступность) — это соотношение количества ЛВ, поступающего в кровь при введении его в различных лекарственных формах (или лекарственных препаратов разных фирм). Если лекарственные препараты демонстрируют схожую биодоступность, они расцениваются как биоэквивалентны.
Общий клиренспрепарата(Cl) — это условный объем крови или ее плазмы, который очищается от ЛВ за единицу времени. Он характеризует скорость элиминации лекарственного препарата из организма. Выражают клиренс в литрах в час или миллилитрах в 1 минуту и рассчитывают по формуле:
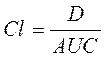
где Сl – общий клиренс; D – доза введенного препарата; AUC – площадь под фармакокинетической кривой.
Элиминация лекарств происходит в почках, печени и других органах (легкие, потовые железы, молочные железы и т.д.). Поэтому выделяют почечный, печеночный и другие виды клиренса, в сумме составляющий общий клиренс:
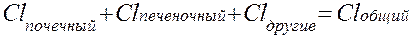
Клиренс в различных органах зависит от скорости кровообращения в органе (Q) и коэффициента экстракции препарата органом (ER):
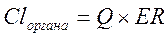
Коэффициент экстракции зависит от количества препарата, поступающего в орган с артериальной кровью (СА) и количества препарата, который выводится из органа с венозной кровью (СV):
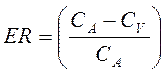
Период полувыведения (Т1/2 или T0,5) — это фармакокинетический показатель времени, в течение которого количество ЛВ в камере или его концентрация в крови уменьшается на 50% .
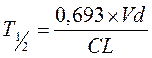 ,
,
где Т1/2 – период полувыведения; 0,693 – коэффициент, который является логарифмом от 2; Vd — объем распределения; Сl — общий клиренс.
Константа элиминации (Кel) — процент уменьшения концентрации ЛВ в крови за единицу времени. Чем больше Кel, тем быстрее ЛВ выводится из крови. Константа элиминации зависит от периода полувыведения:
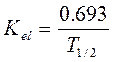
Период полуабсорбции (Т1/2а) — время, необходимое для всасывания половины дозы препарата из места введения в системный кровоток; пропорционален скорости абсорбции (Т1/2а = 0,693/Ка).
Константа абсорбции (Ка) — характеризует скорость всасывания ЛВ в организме человека или животного. Константа абсорбции зависит от периода полувыведения:
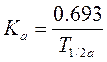
Кажущаяся начальная концентрация (С0) — концентрация, которая была бы в плазме крови при внутривенном пути введения и мгновенном распределении по органам и тканям.
Равновесная концентрация (Сss) — концентрация, которая устанавливается в плазме крови при поступлении препарата в организм с постоянной скоростью. При прерывистом введении препарата через одинаковые промежутки времени в одинаковых дозах выделяют максимальную (Сmax) и минимальную (Сmin) равновесные концентрации.
Дата добавления: 2015-03-03 ; просмотров: 1860 ; ЗАКАЗАТЬ НАПИСАНИЕ РАБОТЫ
Видео:Математика это не ИсламСкачать

Площадь под фармакокинетической кривой
В фармакокинетике понятие «площадь под кривой» имеет особое значение. Оно представляет собой область под линией, нанесенной на график концентрации медицинского препарата в плазме крови в зависимости от времени. Обычно площадь рассчитывается для периода с момента введения препарата до снижения концентрации в плазме до незначительного уровня. Площадь под кривой демонстрирует общее воздействие активного вещества на организм и позволяет оценивать и сравнивать биодоступность различных медицинских препаратов. Время, когда в крови наблюдается самая высокая концентрация активного
вещества, обозначено как Тмакс, а максимальная концентрация активного вещества обозначена Cмакс.
При изучении биоэквивалентности лекарственных препаратов наиболее важными являются следующие фармакокинетические параметры (рис.1)
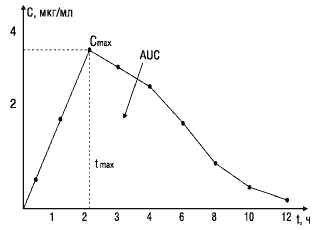 Основные параметры фармакокинетики, используемые при изучении биоэквивалентности лекарственных веществ
Основные параметры фармакокинетики, используемые при изучении биоэквивалентности лекарственных веществ
Cmax — максимум, или пик концентрации лекарственного вещества в крови;
tmax — время достижения максимальной концентрации вещества в плазме крови;
AUC— площадь под фармакокинетической кривой — кривой «концентрация—время» (изменение концентрации активного вещества в плазме или сыворотке крови во времени).
Значение показателя максимальной концентрации вещества можно объяснить с помощью следующего примера. На рис. 2 представлены фармакокинетические кривые двух лекарственных препаратов. Кривая 1 характеризует концентрацию в крови стандартного препарата, кривая 2 — тестируемого. Горизонтальной линией отмечена минимальная эффективная концентрация, при которой данное вещество оказывает терапевтическое действие. Как видно, Сmax тестируемого препарата (кривая 2) не достигает уровня минимальной эффективной концентрации и, следовательно, не оказывает терапевтического действия.
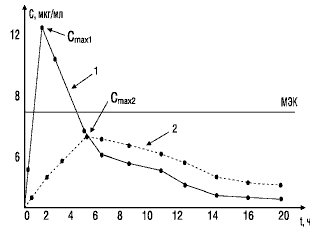
Рис. 2. Сопоставление максимальных концентраций в крови двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; Сmax1, Сmax2 — соответствующие максимальные концентрации сравниваемых препаратов в крови
Второй важный параметр — время достижения максимальной концентрации лекарственного вещества в крови.Этот показатель отражает скорость его всасывания и скорость наступления терапевтического эффекта. На рис. 3 показано, что Сmax стандартного препарата (кривая 1) достигается через 1 ч, а тестируемого (кривая 2) — через 4 ч. Такое различие во времени достижения максимальной концентрации препарата в крови может обусловить изменение клинических показаний к применению данного препарата.
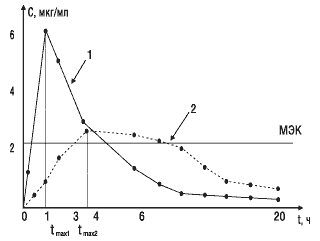
Рис.3. Сопоставление времени достижения максимальной концентрации двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; tmax1, tmax2 — соответствующее время достижения максимальных концентраций сравниваемых препаратов в крови
Третьим важным параметром биоэквивалентности является площадь под фармакокинетической кривой,которая отражает количество вещества, поступившего в кровь после однократного введения препарата. На рис. 3 показано, что две кривые имеют разную форму, разные пики и неодинаковое время достижения максимальной концентрации. Но площади под этими кривыми близки по величине, следовательно, оба препарата обеспечивают поступление в кровь одинакового количества лекарственного вещества.
На рис. 4 представлен другой пример соотношения кривых, отражающих кинетику двух сравниваемых препаратов. Площадь под кривой 1 практически в два раза больше, чем под кривой 2. Обращает на себя внимание то, что максимальная концентрация и время ее достижения схожи у стандартного и тестируемого препаратов. Однако площадь под фармакокинетической кривой у тестируемого препарата в 2 раза меньше за счет более быстрого выведения его из крови. В данном случае можно ожидать уменьшения длительности действия лекарственного препарата и снижения его терапевтического эффекта.
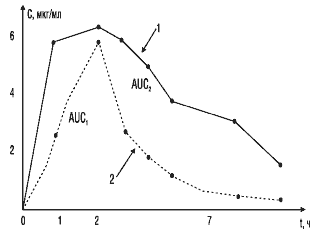
Рис. 4. Сопоставление площадей под фармакокинетическими кривыми двух препаратов: 1 — стандартный препарат; 2 — тестируемый препарат; AUC1, AUC2 — соответствующие площади под кривыми «концентрация—время» сравниваемых препаратов
Таким образом, два препарата считают биоэквивалентными, если они имеют схожие фармакокинетические показатели. По регламенту ВОЗ (1994, 1996) и ЕС (1992) их различие не должно превышать 20%.
Необходимо отметить, что планировать и проводить исследование по определению биоэквивалентности должен коллектив специалистов различных профилей: клинические фармакологи, врачи-клиницисты, биохимики, химики-аналитики, биостатистики. Все этапы проведения исследования должны быть тщательно описаны, проанализированы и представлены в подробном отчете.
Внедрение определения биоэквивалентности как метода позволяет сделать обоснованное заключение о качестве, эффективности и безопасности сравниваемых препаратов на основании меньшего объема первичной информации и в более сжатые сроки, чем при проведении клинических испытаний.
В настоящее время уже существуют регламенты изучения биоэквивалентности ВОЗ (1996), ЕС (1992), Российской Федерации (1995). В них изложены основные обоснования необходимости проведения таких исследований.
Так, изучение биоэквивалентности проводят, если:
· существует риск отсутствия биоэквивалентности и/или
· существует риск снижения фармакотерапевтического действия и клинической безопасности препарата.
Например, обязательно оценивают:
· препараты для лечения состояний, при которых необходим гарантированный терапевтический эффект;
· препараты с узким терапевтическим коридором безопасности;
· препараты, фармакокинетика которых осложнена снижением абсорбции менее 70% или с высокой элиминацией (более 70%);
· препараты с неудовлетворительными физико-химическими свойствами (низкая растворимость, нестабильность, полиморфизм);
· препараты с документированно подтвержденным существованием проблем биодоступности.
В настоящее время в Украине достаточно широко используют высокоэффективные методы для определения фармакокинетических параметров. Расширение имеющейся материально-технической базы, разработка регламента проведения исследований биоэквивалентности лекарственных веществ, подготовка специалистов в этой области позволят решить актуальную задачу по оценке эффективности и безопасности генерических препаратов отечественного и зарубежного производства.
В.И. Мальцев, А.П. Викторов,
БИЛЕТ 8
Изменение эффекта лекарственных средств при повторном применении
В клинической практике лечение пациента редко ограничивается однократным приемом лекарства. При повторном применении препаратов фармакологический эффект может нарастать или снижаться в связи с изменением чувствительности рецепторов к ним или изменением фармакокинетики. Нарастание специфического фармакологического действия при повторных введениях одного и того же лекарственного вещества называют кумуляцией. При материальной кумуляции нарастание эффекта обусловлено постоянным повышением концентрации препарата в крови и тканях из-за медленного его метаболизма и выведения. Это может быть причиной появления токсических эффектов при повторном применении терапевтических доз препарата. Опасность материальной кумуляции возрастает при нарушении функции печени и почек. Например, сердечные гликозиды группы наперстянки применяют для лечения сердечной недостаточности, которая нередко сопровождается патологией печени. В этих условиях биотрансформация препаратов замедляется и проявляется материальная кумуляция: сначала нарастание терапевтического эффекта, затем интоксикация Для предотвращения кумуляции необходимо проводить коррекцию доз (снижать) и интервалов между приемами препарата (увеличивать).
При функциональной кумуляции лекарство вызывает в организме труднообратимые или необратимые изменения, сохраняется следовая реакция, в результате повторное введение препарата может усилить эти изменения. Возникает скачкообразное усиление эффекта, хотя концентрация лекарства в крови и клетках соответствует вводимой дозе. Примером такого вида кумуляции может служить действие этилового спирта на больных алкоголизмом: синдром «белой горячки» может развиться от «обычной» дозы спирта, дипсомания (неодолимая тяга к алкоголю) провоцируется малой дозой алкоголя. Функциональная кумуляция сохраняется иногда пожизненно.
К лекарствам организм при первичном контакте может сенсибилизироваться, и тогда повторное их введение вызовет аллергические реакции (от греческого allos — другой, не специфический, ergon — действие), свидетельствующие об иммунологической несовместимости организма пациента с определенной группой химических веществ. Препараты такой структуры пациенту назначать опасно.
При повторном введении лекарств может наблюдаться и снижение специфического эффекта. Быстро возникающее (и также быстро исчезающее) понижение чувствительности организма к лекарству может быть обусловлено истощением метаболита, субстрата, через который оно реализует свой эффект — это явление называется тахифилаксией (от греческого tachys -быстрый, phylaxis -охрана). Она возникает к некоторым сосудосуживающим средствам (например, эфедрин), дыхательным аналептикам из группы Н-холиномиметиков (например, цититон). Первоначальный эффект восстановится, когда нормализуется уровень биосубстрата, с которым лекарство образует комплекс.
Ко многим лекарствам (снотворным, болеутоляющим, слабительным) при повторном применении развивается привыкание или толерантность (устойчивость). В этом случае повторное назначение терапевтических доз дает все меньший эффект. Наиболее вероятная причина — изменения фармакокинетики: уменьшение всасывания, увеличение скорости биотрансформации (индукции ферментов) и выведения. Для получения первоначального эффекта необходимо повысить дозу.
Длительное применение препаратов, действующих на центральную нервную систему и вызывающих эйфорию (эу — хорошо, фора — чувствую). может сопровождаться развитием лекарственной зависимости. Отмена препарата приводит к состоянию абстиненции (синдром «лишения»), поскольку при повторном контакте препарат, вызывающий зависимость, вероятно, включается в метаболизм нервной ткани. Различают психическую и физическую зависимость. При психической лекарственной зависимости отмена препарата вызывает эмоциональный дискомфорт и желание принять препарат становится самоцелью. При физической лекарственной зависимости наряду с психическими изменениями наблюдается нарушение функции различных органов и систем. Тяжелое состояние (абстиненция), развивающееся у пациента, не получающего соответствующий наркотик, заставляет его принимать лекарство уже не в соответствии с его истинным назначением. Причем параллельно с лекарственной зависимостью часто развивается привыкание (толерантность), и для снятия абстиненции требуются все большие дозы наркотика. С увеличением дозы усугубляется и последующий абстинентный синдром, усиливаются нарушения психических и соматических функций, развиваются хроническое отравление организма и моральная деградация личности.
Профилактика и лечение наркомании — сложная медико-социальная проблема. Лечение больных, страдающих лекарственной зависимостью, проводят в специальных учреждениях под наблюдением врачей-наркологов.
Если при психической зависимости наркотик можно отменить сразу, то при физической зависимости одномоментная отмена препарата может привести к тяжелым осложнениям, вплоть до смертельных.
Лекарственную зависимость вызывают наркотические анальгетики, кокаин, спирт этиловый, снотворные, транквилизаторы, психостимуляторы, галлюциногены и другие психотропныепрепараты.
БИЛЕТ 7
Виды действия ЛС
ДЕЙСТВИЕ ПРЕПАРАТОВ МОЖЕТ БЫТЬ:
1. МЕСТНОЕ и РЕЗОРБТИВНОЕ.
МЕСТНОЕ действие лекарственных средств развивается в месте их применения. Например, обезболивающее действие местных анестетиков новокаина, лидокаина и др.
РЕЗОРБТИВНОЕ действие препаратов развивается после всасывания в кровь и проникновения до органа — мишени через гистогематические барьеры (например: сердечные гликозиды: дигоксин, коргликон и др. оказывают свой основной положительный инотропный эффект на мышцу сердца в результате резорбтивного действия).
2. ПРЯМОЕ и НЕПРЯМОЕ (в части случаев рефлекторное действие).
Прямое действие лекарственных средств развивается непосредственно в органе — мишени. Это действие может быть местным, например: местный анестетик лидокаин оказывает местный обезболивающий эффект, и резорбтивным, например, местный анестетик лидокаин применяется в качестве антиаритмического препарата, для того, чтобы лидокаин оказал лечебный эффект при желудочковых тахиаритмиях сердца, лидокаин должен всосаться в кровь и пройти гисто- гематические барьеры до очага аритмии в ткани сердца.
Непрямое действие можно рассмотреть на примере действия сердечных гликозидов (дигоксина, строфантина и др.). Дигоксин оказывает стимулирующее влияние на сократимость сердечной мышцы, в результате увеличивается сердечный выброс. Скорость кровотока возрастает и увеличивается перфузия (кровоток) в почках. Это приводит к повышению уровня диуреза (количество мочи увеличивается). Таким образом, дигоксин косвенно увеличивает диурез через стимуляцию сократимости миокарда.
Рефлекторное действие лекарственных средств развивается в том случае, когда в одном месте организма препарат изменяет активность рецепторов, и в результате этого эффекта в другом месте организма изменяется функция органа (например: нашатырный спирт, возбуждая рецепторы слизистой носовой полости приводит к возбуждению клеток дыхательного центра головного мозга, в результате повышается частота и глубина дыхания).
- ИЗБИРАТЕЛЬНОЕ и НЕИЗБИРАТЕЛЬНОЕ.
Избирательное (элективное) действие лекарственных
средств осуществляется путем влияния на определенные рецепторы (например: празозин блокирует преимущественно Л1|-адренорецепторы) или ЛС могут накапливаться в определенном органе и оказывать присущий им эффект (например: йод избирательно накапливается в щитовидной железе, и там изменяет функцию этого органа). В клинической практике считается, что чем выше избирательность действия ЛС, тем меньше токсичность и выраженность отрицательных побочных реакций.
Неизбирательное действие препаратов, термин противоположный избирательному эффекту (например: наркозное средство фторотан неизбирательно блокирует практически все виды рецепторных образований в организме, преимущественно в нервной системе, что приводит к бессознательному состоянию, то есть наркозу).
Обратимое действие ЛС обусловлено непрочностью химических взаимодействий с рецепторными образованиями или ферментами (водородные связи и др.; например: антихо- линэстеразное средство обратимого типа действия — прозерин). Необратимое действие наступает, когда с рецепторами или ферментами ЛС связывается прочно (ковалентные связи; например: антихолинэстеразное средство необратимого типа действия — армин). 5. ГЛАВНОЕ и ПОБОЧНОЕ. Главное действие ЛС — это эффект препарата, направленный на лечение основного заболевания (например: доксазозин — альфа-1-адреноблокатор применяется для лечения гипертонической болезни). Побочное действие — это эффекты препарата не направленные на лечение основного заболевания. Побочное действие может быть ПОЛОЖИТЕЛЬНЫМ (например: доксазозин при курсовом лечении гипертонической болезни тормозит рост предстательной железы и нормализует тонус сфинктера мочевого пузыря, и, следовательно, может применяться при аденоме предстательной железы и нарушениях мочеиспускания) и ОТРИЦАТЕЛЬНЫМ (например: доксазозин может вызывать преходящую тахикардию при лечении гипертонической болезни, а также часто регистрируют синдром отмены). АГОНИСТЫ — лекарственные средства, возбуждающие рецепторные образования. Например: орциприналина сульфат (асмопент) стимулирует р 2 -адренорецепторы бронхов и приводит к расширению просвета бронхов. АНТАГОНИСТЫ — лекарственные средства, блокирующие возбуждение рецепторов (метопролол блокирует бета-1-адренорецепторы в мышце сердца и уменьшает силу сердечных сокращении). АГОНИСТЫ-АНТАГОНИСТЫ — лекарственные средства, обладающие свойствами как возбуждать, так и угнетать рецепторные образования. Например: пиндолол (вискен) блокирует бета-1- и бета-2-адренорецепторы. Однако пиндолол обладает так называемой «внутренней симпатомиметической активностью», то есть препарат, блокируя бета- адренорецепторы и препятствуя определенное время воздействию медиатора на эти рецепторы, оказывает и некоторое стимулирующее влияние на те же бета- адренорецепторы. Дозы лекарственных средств
БИЛЕТ 6
Основное и побочное действие ЛС
Основное действие. Действие, ради которого применяется лекарственное вещество при лечении данного заболевания. Например, фенитоин (дифенин) обладает противосудорожными и антиаритмическими свойствами. У больного эпилепсией основным действием фенитоина является противосудорожное, а у больного с сердечной аритмией, вызванной передозировкой сердечных гликози- дов — антиаритмическое.
Все остальные эффекты лекарственного вещества (кроме основного), которые возникают при его приеме в терапевтических дозах, расцениваются как проявления побочного действия. Эти эффекты часто бывают неблагоприятными (отрицательными) (см. главу 5). Например, ацетилсалициловая кислота может вызвать изъязвление слизистой оболочки желудка, антибиотики из группы аминогликозидов (канамицин, гентамицин и др.) — нарушать слух. Отрицательное побочное действие часто является причиной ограничения применения того или иного лекарственного вещества и даже исключения его из списка лекарственных препаратов.
екарственные средства назначают для достижения определенного фармакотерапевтического эффекта (обезболивания, снижения АД и т. д.). Все это является проявлением основного действия препаратов. Однако наряду с желательными эффектами любые лекарственные вещества могут оказывать и неблагоприятное воздействие (побочные реакции неаллергической природы, аллергические реакции, токсические и другие эффекты).
К проявлениям побочного действия неаллергического происхождения относят только те эффекты, которые возникают при применении веществ в терапевтических дозах и составляют спектр их фармакологического действия. Так, фенобарбитал при использовании в качестве противоэпилептического препарата может быть причиной сонливости.
Побочное действие может быть первичным или вторичным. Первичное действие возникает как прямое следствие влияния данного препарата на определенный субстрат (тошнота и рвота при раздражающем действии на слизистую оболочку желудка). Вторичное действие относится к косвенно возникающим неблагоприятным влияниям (гиповитаминоз при подавлении кишечной флоры антибиотиками).
Аллергические реакции возникают независимо от дозы вводимого вещества и подразделяются на 4 типа:
- тип I (немедленная аллергия) — проявляется крапивницей, сосудистым отеком, ринитом, бронхоспазмом и анафилактическим шоком. Такая реакция возможна при применении пенициллинов и сульфаниламидов;
- тип II — через систему комплемента IgG и IgM взаимодействуют с клетками крови, вызывая их лизис. Так, метилдофа может вызывать гемолитическую анемию, а анальгин — агранулоцитоз;
- тип III — комплекс антиген-антитело-комплемент повреждает сосудистый эндотелий. Возникает сывороточная болезнь (крапивница, артралгия, артрит, лимфаденопатия, лихорадка). Подобную реакцию вызывают пенициллины, сульфаниламиды, йодиды;
- тип IV — возникает при местном нанесении вещества и проявляется контактным дерматитом.
Идиосинкразия — атипичная реакция организма на лекарственное вещество. Является одним из видов неблагоприятной реакции на вещества. Токсическое действие наблюдаются при передозировке, отравлениях или накоплении токсических веществ при нарушении их метаболизма (патология печени и почек).
Тератогенное действие — отрицательное действие лекарственных веществ на плод и эмбрион, приводящее к рождению детей с аномалиями. Наиболее опасное время беременности — первый триместр (3-8-я недели).
Эмбриотоксическое действие — неблагоприятное воздействие на эмбрион до 12 недель беременности, не связанное с нарушением органогенеза (не относится к тератогенному действию). Фетотоксическое действие — неблагоприятное воздействие на эмбрион после 12 недель беременности, также не связанное с нарушением органогенеза.
Мутагенность — способность повреждать генетический аппарат клетки и вызывать мутации. Канцерогенность — способность лекарственных веществ вызывать развитие злокачественных опухолей.
Лекарственная несовместимость — неблагоприятные эффекты лекарственных веществ, возникшие при их сочетанном действии.
БИЛЕТ 5
Пути выведения лекарственных средств из организма. Клиническое значение.
Л.В. выводятся из организма либо в неизменном виде, либо в виде метаболитов и конъюгатов.
1. Основной путь – выведение через почки с мочой
А) полярные, водорастворимые вещества легко фильтруются в клубочках и быстро выводятся;
Б) неполярные, липофильные вещества после фильтрации возвращаются опять в кровь. Затем поступают в печень и подвергаются БТ. Образовавшиеся водорастворимые метаболиты выводятся через почки. Полярные вещества плохо реабсорбируются, а неполярные вещества хорошо
Через почки выводится только свободная фракция лекарства, не связанная с белками. Имеет значение рН мочи, см.выше.
При заболевании почек их способность выводить лекарственные вещества снижается.
2. Через ЖКТ
а) через кишечник – выводятся вещества, которые не всасываются в ЖКТ (фталазол, активизированный уголь);
 б) с желчью – всосавшиеся вещества поступают в печень желчь просвет кишечника (САП, противотуберкулезные, некоторые АБ).
б) с желчью – всосавшиеся вещества поступают в печень желчь просвет кишечника (САП, противотуберкулезные, некоторые АБ).
Некоторые вещества выделяющиеся с желчью, могут повторно всасываться в кровь и в дальнейшем вновь выделяться в кишечник и так несколько раз (кишечно-печеночная циркуляция)
в) через эпителий ЖКТ может идти секреция веществ из плазмы крови в просвет кишечника (тяжелые металлы, сенна)
3. Через легкие– газообразные или летучие вещества (средства для наркоза,NH4OH,C2Н5ОН).
4. Через секреты желез
· со слюной – эритромецин, иодиды, ампициллин, Bi;
· с потом – пенициллины, гриозеофульвин;
· со слезами – рифампицин;
· через кожу – соли тяжелых металлов;
· с молоком матери – седативные, снотворные, анальгетики, алкоголь, никотин, противомикробные средства, глюкокортикоиды и др
БИЛЕТ 4
Механизмы всасывания ЛС.
ОСНОВНЫЕ МЕХАНИЗМЫ ВСАСЫВАНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ (ЛС)
Всасывание – это процесс поступления ЛС из места введения в кровь. Всасывание лекарственного вещества зависит от пути введения его в организм, лекарственной формы, физико-химических свойств (растворимости в липидах или гидрофильности вещества), а также от интенсивности кровотока в месте введения.
ЛС, принятые перорально, подвергаются всасыванию, проходя через слизистую оболочку желудочно-кишечного тракта, что определяется их растворимостью в липидах и степенью ионизации. Различают 4 основные механизма всасывания: диффузия, фильтрация, активный транспорт, пиноцитоз.
Пассивная диффузия осуществляется через клеточную мембрану. Всасывание происходит до тех пор, пока концентрация лекарственного вещества по обе стороны биомембраны не сравняется. Подобным образом всасываются липофильные вещества (например, барбитураты, бензодиазепины, метопролол и др.), причем чем выше их липофильность, тем активнее их проникновение через клеточную мембрану. Пассивная диффузия веществ идет без затраты энергии по градиенту концентрации.
Облегченная диффузия – это транспорт лекарственных веществ через биологические мембраны с участием молекул специфических переносчиков. При этом перенос лекарства осуществляется также по градиенту концентрации, но скорость переноса при этом значительно выше. Например, таким образом всасывается цианокобаламин. В осуществлении его диффузии участвует специфический белок – гастромукопротеид (внутренний фактор Кастла), образующийся в желудке. Если продукция этого соединения нарушена, то снижается всасывание цианокобаламина и, как следствие этого, развивается пернициозная анемия.
Фильтрация осуществляется через поры клеточных мембран. Этот механизм пассивного всасывания идет без затраты энергии и осуществляется по градиенту концентрации. Характерен для гидрофильных веществ (например, атенолол, лизиноприл и др.), а также ионизированных соединений.
Активный транспорт осуществляется с участием специфических транспортных систем клеточных мембран. В отличие от пассивной диффузии и фильтрации активный транспорт процесс энергозатратный и способен осуществляться против градиента концентрации. В данном случае несколько веществ могут конкурировать за один и тот же транспортный механизм. Способы активного транспорта обладают высокой специфичностью, поскольку сформировались в процессе длительной эволюции организма для обеспечения его физиологических потребностей. Именно эти механизмы являются основными для осуществления доставки в клетки питательных веществ и выведения продуктов обмена.
Пиноцитоз (корпускулярная абсорбция или пенсорбция) представляет также разновидность всасывания с затратой энергии, осуществление которого возможно против градиента концентрации. При этом происходит захват лекарственного вещества и инвагинация клеточной мембраны с образованием вакуоли, которая направляется к противоположной стороне клетки, где происходит экзоцитоз с высвобождением лекарственного соединения.
БИЛЕТ 3
Фармакокинетика– это раздел фармакологии, который изучает различные этапы прохождения распределение по органам и тканям, биотрансформация, выведение ЛС из организма.
Фармакокинетика лекарственных средств в организме представлены этапами, связанными с:
· Проникновением – регулируются процессами диффузии (смешивание различных веществ до однородного состояния), фильтрации и активного транспорта (движение некоторых медикаментозных веществ в клетки и из них против концентрационного реагента)
· Всасыванием – действие, связанное с поступлением медикамента в кровь. Определяется лекарственной формой, растворимостью, кровотоком в месте введения
· Распределение – определяется растворимостью препарата в липидах, связью между белками крови и кровотоком
· Биотрансформацией – процесс биохимических и физико-химических действий. Как правило, на биотрансформацию влияют заболевания печени, особенности питания, возраст и др.
· Существуют разные пути введения лекарственного средства в организм. Их можно разделить на 2 большие группы: энтеральный (через желудочно-кишечный тракт), парентеральный (минуя желудочно-кишечный тракт). К энтеральным путям введения относят: пероральный (peros– через рот), сублингвальный (под язык), через зонд в желудок и двенадцатиперстную кишку, ректальный (через прямую кишку). К парентеральным путям введения относятся: накожный, внутрикожный, подкожный, внутримышечный, внутривенный, внутриартериальный, внутрисердечный, под оболочки мозга, ингаляционный, интрастернальный (в грудину). Каждый из путей введения имеет свои преимущества и недостатки.
· Самый распространенный путь введения – это через рот (пероральный). Этот путь удобный, простой, не требуется стерильность препаратов. Всасывание лекарственного вещества идет частично в желудке, частично в кишечнике. Однако некоторые лекарственные вещества могут разрушаться под действием желудочного сока. В этом случае лекарственное вещество помещают в капсулы, которые не разрушаются желудочным соком. Под языком лекарственное средство всасывается быстро, минует печень и не вступает в контакт с содержимым желудка и кишечника (Нитроглицерин). При ректальном способе введения (суппозитории, клизмы) лекарственное вещество быстро всасывается, частично минуя печень. Однако, далеко не все препараты хорошо всасываются из слизистой прямой кишки, а некоторые препараты могут раздражать слизистые оболочки.
· Из парентеральных путей введения чаще используют: под кожу, внутримышечный, внутривенный. Быстрый эффект наступает при внутривенном пути введения. Однако к трудностям парентеральных способов введения относят: болезненность укола, стерильность препаратов и шприцов, необходимость медицинского персонала для проведения инъекций.
· Поступив в организм, лекарственное вещество должно всосаться. Всасывание (абсорбция) – это процесс поступления лекарственного вещества в кровеносную или лимфатическую систему из места введения. Основные механизмы всасывания: пассивная диффузия, облегченная диффузия, активный транспорт, пиноцитоз. Факторы, влияющие на всасывание лекарственного вещества при приеме внутрь: растворимость, лекарственная форма, pHжелудка и кишечника, активность ферментов желудочно-кишечного тракта, перистальтика желудочно-кишечного тракта, прием пищи, мальабсорбция, дисбактериоз.
· После всасывания лекарственного вещества в кровь оно будет циркулировать там, в «свободной» или «связанной» форме. «Свободная» форма (не связана с белками крови) растворима в водной фазе плазмы крови. Эта форма легко проникает через стенку капилляров в ткани и оказывает фармакологический эффект. «Связанная» форма – это часть лекарственного вещества, которая связана с белками крови (чаще с альбуминами) и неспособна, проникать в ткани. Эта форма представляет собой как бы депо препарата и по мере выведения лекарственного вещества из организма отщепляется от белка и переходит в «свободную» форму. Следовательно: только «свободная» форма лекарственного вещества оказывает фармакологический эффект.
· После всасывания в кровь лекарственное вещество подвергается распределению по органам и тканям. Распределение по органам и тканям чаще всего бывает неравномерным. Степень поступления в ту или иную ткань зависит от разных факторов: от молекулярной массы, от растворимости в воде и липидах, от степени диссоциации; от возраста, пола; от массы жировых депо; от функционального состояния печени, почек, сердца; от способности преодолевать гистогематические барьеры.
· К гистогематическим барьерам относят: капиллярную стенку, гематоэнцефалический барьер, гематоофтальмический барьер, плацентарный барьер. Капилляры легко проницаемы для лекарственных веществ, так как стенка капилляров имеет широкие поры, через которые легко проходят водорастворимые вещества с молекулярной массой не больше инсулина (5 – 6 кДа). А жирорастворимые вещества диффундируют через мембрану клеток.
· Гематоэнцефалический барьер – представляет собой капиллярную стенку, которая является многослойной мембраной (эндотелий, межуточное вещество и глиальные клетки головного и спинного мозга). Такая мембрана лишена пор. Через гематоэнцефалический барьер легко проникают липофильные вещества путем простой диффузии (например, тиопентал натрия – наркозное средство). Для полярных соединений (пенициллины, миорелаксанты) гематоэнцефалический барьер не проницаем. Гематоэнцефалический барьер гипоталамуса, гипофиза отличается повышенной проницаемостью для лекарственных веществ. Проницаемость гематоэнцефалического барьера повышается при менингите, арахноидите, гипоксии, черепно-мозговых травмах. Некоторые лекарственные препараты (кофеин, эуфиллин, лидаза) повышают проницаемость гематоэнцефалического барьера.
· Гематоофтальмический барьер отделяет кровь капилляров от внутриглазной жидкости в камерах глаза. В камеры глаза хорошо проходят липофильные препараты.
· Плацентарный барьер разделяет кровообращение матери и плода. На ранних стадиях беременности наблюдается большая порозность этого барьера и многие лекарства легко проникают в плод. Затем этот барьер «укрепляется» и приобретает свойства липидной мембраны. Но с 33 – 35-й недели беременности истончается плацента и значительно повышается проницаемость плацентарного барьера. Это создает опасную ситуацию для плода. Не проникают через плацентарный барьер крупномолекулярные вещества (инсулин, полиглюкин), а также гидрофильные ионизированные молекулы: миорелаксанты, ганглиблокаторы.
· Следующий этап фармакокинетики – это элиминация лекарственного вещества. Элиминация (от латинского eliminatum– удалять) – удаление лекарств из организма путем биотрансформации и экскреции.
· Биотрансформация – это метаболическое превращение лекарств, в результате которых они приобретают полярные группы, то есть уменьшается растворимость в липидах и возрастает растворимость в воде. Полярные метаболиты пригодны к удалению из организма. Для примера хочу сказать, что если бы не было метаболизма, то одна терапевтическая доза снотворного средства этаминала могла бы находиться в организме 100 лет. Биотрансформация лекарств чаще всего (90 – 95%) происходит в печени, реже в слизистой оболочке кишечника, почках, легких, коже, в крови. Наиболее изучен метаболизм лекарств в печени. Метаболизм в печени происходит: либо в эндоплазматическом ретикулуме гепатоцитов с помощью микросомальных оксидаз смешанной функции либо вне эндоплазматического ретикулума (в митохондриях) с помощью немикросомальных ферментов
Фармакодинамика
Фармакодинамика (от греческого «pharmakon» — лекарство, «dynamikos» — сильный) — раздел фармакологии, рассматривающий совокупность эффективности лекарственных средств, механизмы их действия на организм человека, а так же взаимосвязь концентрации лекарственных веществ с достигнутым действием их на организм.
Так достигаемые эффекты отражаются в простом уравнения L + R L*R, где L — это лекарство, а R — это рецептор (место приложения действия). В этом и заключается определение фармакодинамики.
Методы для изучения фармакодинамики:
Ключевой момент в методике изучения это следующие важные свойства:
а) высокая чувствительность — способность выявлять большую часть отклонений от исходного состояния, и динамику положительных изменения в организме.
б) высокая специфичность — способность редко давать «ложноположительные» результаты.
в) высокая воспроизводимость — способность стабильно отображать характеристики состояния больных при повторных исследованиях у одних и тех же пациентов и одинаковых условиях при отсутствии какой-либо динамики в состоянии этих больных по другим клиническим данным.
Видео:Фармакология. Фармакокинетика (простым языком).Скачать

ОСНОВНЫЕ ФАРМАКОКИНЕТИЧЕСКИЕ ПОКАЗАТЕЛИ
Фармакокинетика наиболее интенсивно начала развиться в последние четыре десятилетия, широко используя физико-химические, биохимические, математические, статистические методы исследования для установления не только судьбы препарата в организме, но также общих закономерностей взаимодействия между лекарством и тканевыми мишенями.
При определении фармакокинетических показателей организм человека или экспериментального животного рассматривается в качестве особой биологической среды, где происходит распределение лекарственных средств в органах, тканях, клетках, субклеточных структурах, их биотрансформация, а также осуществляется взаимодействие медикаментов с тканевыми рецепторами.
Одним из основных показателей, определяющих эффективность лекарства — концентрация медикамента в области рецептора или ткани, где происходит взаимодействие между препаратом и организмом. Определить концентрацию медикамента в организме человека в конкретном органе или в ткани практически невозможно. Для определения фармакокинетических параметров регистрируют количество медикамента в крови, принимая во внимания положение, что в большинстве случаев имеется прямая корреляция между концентрацией препарата в крови и его количеством в области рецептора. На основании полученных данных строят график — фармакокинетическую кривую, где на оси ординат отмечают концентрацию препарата в плазме крови, а на оси абсцисс – время исследования.
Выделяют основные параметры и термины фармакокинетики:
Камера – условное понятие в фармакокинетике, под которым понимают пространство, характеризующееся определенным объемом и концентрацией медикамента в этом пространстве. Понятие камера не отражает какое-либо анатомическое пространство. Это единица формализованной фармакокинетической системы, принятая в мире для математического моделирования процессов, происходящих в организме при взаимодействии лекарства с организмом.
Различают центральную камеру, за которую принимают кровь и хорошо кровоснабжаемые органы: сердце, почки, легкие, эндокринные органы, печень, кишечник. К периферической камере относят менее интенсивно кровоснабжаемые органы и ткани: кожа и подкожная клетчатка, мышцы, жировая ткань и др.
Условно простейшая модель взаимодействия лекарственного средства с организмом рассматривается как однокамерная или многокамерная модель, характеризующаяся концентрацией лекарства (Сл) и объемом распределения (Vp).
Концентрация лекарства (Сл) –количество препарата в определенном объёме крови в конкретный момент после введения лекарства в организм. Концентрацию лекарства в организме определяют спектрофотометрическим, хроматографическим, полярографическим, ферментным, радиоиммунным и другими методами, выражая в мг/л, мкг/мл, мм/л или в %.
Динамика концентрации медикамента в организме зависит от пути введения, дозы, физико-химических свойств, длительности действия препарата и др. Наиболее простая фармакокинетическая модель – однокамерная модель, где организм представляется в виде гомогенной единой камеры. Однокамерная модель применима для определения концентрации лекарства в крови, плазме и сыворотке, а также в моче.
Фармакокинетические процессы в наибольшей степени соответствуют процессам, происходящим в организме, в случае двух- или трехкамерной модели.
Объем распределения (Vd) – (кажущийся, гипотетический объем распределения препарата) — условный объем жидкости, необходимый для равномерного распределения (растворения) введенной дозы лекарства до концентрации, обнаруживаемой в крови в момент исследования; что выражается в литрах на килограмм массы тела (л/кг).
Объем распределения медикамента в определенной мере характеризует степень проникновения лекарства из плазмы крови и внеклеточной жидкости в ткани и создания депо лекарственного препарата в органах. Например, для антибиотиков группы аминогликозидов, мало растворимых в липидах, объем распределения близок к объему внеклеточной жидкости, а для хорошо растворимых в липидах тетрациклинов—значительно выше.
Если препарат активно проникает в биологические органы и ткани, то это свидетельствует о высоком значении объёма распределения. Данный фармакокинетический показатель зависит от путей введения, дозы, физико-химических свойств медикамента (растворимость в жирах и воде, степень ионизации и полярности, молекулярная масса), а также возраста, пола, количества жидкости в организме, патологического состояния организма (заболеваний печени, почек, сердечно-сосудистой системы, кишечника).
Площадь под кинетической кривой концентрация-время(area under curve, AUC). AUC при линейной кинетической кривой (линейная зависимость) пропорциональна количеству медикамента, находящемуся в системном кровотоке.
Биодоступность (F) определяется относительным количеством лекарства, которое освобождается из лекарственной формы, поступает в общий круг кровообращения и взаимодействует с тканевыми рецепторами, выраженное в процентном отношении. Биодоступность определяют по формуле:
AUC (в/м или per os)
AUC (в/м или per os) D (в/в)
AUC (в/в) D (в/м или per os)
Биодоступность зависит от химической структуры медикамента, технологии приготовления лекарственной формы и от степени абсорбции лекарства в кровь из пищеварительного тракта при энтеральном поступлении, биотрансформации при первом прохождении (пассаже) через печень, скорости рассасывания при парентеральном введении медикамента. Биодоступность препарата при введении непосредственно в кровь принимают за 100% и при других путях поступления – выражают в %.
Биоэквивалетность(сравнительная биодоступность) – соотношение количества медикамента, поступающего в кровь при применении его в той или иной лекарственной форме или в лекарственных препаратах, выпускаемых различными фирмами. Изучение биоэквивалентности позволяет сравнивать препараты в клинической практике, что очень важно для определения эффективности медикаментов различных производителей.
Биофаза – участок непосредственного взаимодействия медикамента с рецептором или тканевой структурой, включая клеточную оболочку и внешнюю, митохондриальную, эндоплазматическую, лизосомальную, рибосомальную мембраны.
Общий клиренс — условный объем плазмы или крови, освобождающийся («очищающийся») от лекарственного средства за единицу времени; выражается в объемных единицах (л/мин, мл/мин).
Общий клиренс вычисляют по формуле:
Где С — общий клиренс, D – доза введенного препарата, AUC – площадь под фармакокинетической кривой.
Различают почечный клиренс – скорость выведения медикамента с мочой, печеночный клиренс – скорость инактивации лекарства в печени, и жёлчный клиренс – скорость выведения препарата с жёлчью.
Почечный клиренс отражает элиминацию препарата из организма и рассчитывается путем деления количества лекарства, экскретируемого с мочой на концентрацию лекарства в плазме крови:
где Смоче — концентрация лекарства в моче;
Сплазме — концентрация лекарства в плазме;
V — объем выделенной мочи.
Для определения клиренса применяют неметаболизируемый лекарственный препарат, который полностью выводится из организма в не измененном виде. В этом случае величина клиренса характеризует функциональную активность органов выделения. При нормальной функции органов выделения, в частности в исследованиях на экспериментальных животных, величина клиренса неизмененного лекарственного средства характеризует степень его метаболических превращений в организме.
Период полувыведения — Т½ (период полуэлиминации) — фармакокинетический показатель, характеризующий время, в течение которого количество медикамента в теоретической камере или его концентрация в исследуемой ткани, в частности в крови, уменьшается на 50%.
Где, Т½ — период полувыведения,0,693 – коэффициент, представляющий собой натуральный логарифм из 2, Vd – объем распределения, С – общий клиренс.
Принято считать, что за один период полувыведения выводится 50% введенного медикамента, за два периода – 75%, за три — 90%.
Из приведенной формулы вытекает, что период полувыведения не может полностью отразить процесс выделения препарата из организма. Это обусловлено тем, что при определенных условиях (уменьшение кровотока в органах, особенно в почках, плохом проникновении медикамента через мембраны) возможно снижение объема распределения и клиренса препарата, вследствие этого период полувыведения не претерпевает существенных изменений, что может повлечь за собой накопление в крови лекарства в токсических концентрациях.
Кроме периода полувыведения определяют также полупериод элиминации (t 1|2 β) и полупериод полураспределения (t 1|2). Чем меньше константа элиминации препарата (Кэл), тем дольше период полувыведения.
Константа элиминации(Кэл) — процент снижения концентрации лекарства в крови в единицу времени. Чем выше Кэл, тем быстрее лекарственное средство удаляется из крови. Если, например, Кэл двух лекарств равны 0,2 ч -1 и 0,025 ч -1 , то это значит, что ежечасно концентрация в крови этих веществ снижается соответственно на 25 % и на 2,5 % одноразовой исходной концентрации.
Глава 3. Фармакодинамика лекарственных средств
Фармакодинамика(греч. pharmacon – лекарство, яд, зелье; dynamis – сила) – комплекс изменений в организме под влиянием лекарственных средств. Основная цель фармакодинамики – изучение механизма действие лекарств и, в первую очередь, первичной фармакологической реакции.
Фармакологический эффект или действие препарата это изменение метаболизма и функции клеток, органов или систем организма, возникающее под влиянием лекарственного средства. В научной литературе такое взаимодействие между лекарственным средством и организмом, которое характеризуется физико–химическими, биохимическими, функциональными изменениями называют первичной фармакологической реакцией. Особенности первичной фармакологической реакции определяются факторами со стороны лекарства: источниками получения, физико-химическими свойствами, химической структурой, дозой (концентрацией) препарата, лекарственной формой, путем, длительностью, последовательностью введения лекарственных средств, а также их сочетаниями.
Кроме того, на первичную фармакологическую реакцию влияют факторы со стороны организма (пол, возраст, масса, функциональное и патологическое состояние организма, генетические особенности, биологические ритмы) и внешней среды. Таким образом, правильное представление о первичной фармакологической реакции лекарственного средства можно составить только при комплексной оценке взаимодействия медикамента с организмом и окружающей средой. Механизмом действияназывают способ, которым реализуется первичная фармакологическая реакция.
Фармакологический эффект это результат взаимодействия между фармпрепаратом и организмом, начинающийся с влияния лекарственного вещества на клеточные мишени или рецепторы. Затем происходят изменения по типу торможения (угнетения) или возбуждения (стимуляция) как функции, так и обмена веществ в тканях и органах. Рассматривая взаимодействие лекарства с клеткой, необходимо выделить понятие первичной фармакологической реакции. Механизмы последней основываются на усилении или угнетении биофизических, химических, биохимических и физиологических процессов в клетках. Чтобы вызвать фармакологический эффект, лекарственное вещество должно вступить в связь с биомолекулами клеток организма. Характер такого влияния предопределяет механизм действия лекарств. Это сложный процесс. Существует несколько гипотез, объясняющих механизм действия лекарств.
Согласно оккупационной теории, предложенной А.Clark, эффект, вызванный лекарственным средством, пропорционален величине поверхности рецепторов, занятой молекулами этого соединения. Максимальный эффект развивается тогда, когда все рецепторы заняты лекарством. Однако действие медиаторов (ацетилхолин, норадреналин) и некоторых других физиологически активных лекарственных веществ на основании данной гипотезы объяснить нельзя. Подобного типа вещества стали называть агонистами, имея в виду противопоставление их антагонистам. Они способны оказывать стимулирующее действие на естественные физиологические функции.
Сложная оккупационная теория разрабатывалась E. Ariens с целью дополнить теорию А. Clark. Выдвинуто предположение о том, что лекарственное вещество обладает двумя независимыми характеристиками: сродством к рецептору и внутренней или собственной активностью. Для антагонистов (ингибиторов) достаточно иметь сродство к рецептору, то есть прочно фиксироваться на его активных центрах. Агонисты должны обладать как сродством к рецепторам, так и внутренней активностью. Последнее свойство позволяет такому веществу вызывать положительную фармакологическую реакцию. Образовавшийся комплекс стимулирует или угнетает функциональное состояние рецептора, клетки, органа и организма в целом. Лекарственное средство образует комплекс с определенной биохимической структурой организма, например рецептором (бета-адреноблокаторы – анаприлин, метопролол) или ферментом (антихолинэстеразный препарат – фосфакол), в результате чего активность рецептора временно угнетается, а активность фермента не восстанавливается, либо восстанавливается не полностью. В этом случае необходим синтез нового фермента.
Попытка дальнейшего углубления вышеназванных положений проводилась R. Stephenson. Автор доказывает, что максимальный эффект достигается тогда, когда лекарственное вещество занимает небольшую часть рецепторов, ибо интенсивность биологической реакции не линейно зависит от числа занятых рецепторов. В таких случаях средство называют высокоэффективным.
От всех предыдущих теорий отличается гипотеза о механизме действия лекарственных веществ, выдвинутая W. Paton, согласно которой интенсивность физиологической реакции, вызванной агонистом, пропорциональна скорости взаимодействия препарата (агониста) с рецептором и не зависит от степени насыщения им рецептора. По мнению автора, если лекарственное вещество непродолжительное время задерживается на рецепторе, то является стимулятором (агонистом). Если же фармпрепарат медленно диссоциирует из комплекса с рецептором, то он является антагонистом.
Представляет интерес наблюдение Z. Hurwitz и сотрудников об активизации транспорта Са 2+ при реакции агониста с рецептором. Именно комплекс рецептор – медиатор или формирует поры для Са 2+ , или является переносчиком данного иона через биологическую мембрану. На кафедре фармакологии Национального медицинского университета, проведенные дополнительные исследования в этом направлении показали, что не только кальций, но и магний способны образовывать комплексы с сердечными гликозидами. Вышеуказанные результаты дают основание высказывать предположение о возможной роли сердечных гликозидов в качестве переносчиков ионов кальция.
В основе первичной фармакологической реакции лежит перенос протонов и электронов с одного вещества на другое, осуществляемый несколькими типами химических связей.
Наиболее универсальный тип связей ван-дер-ваальсовые, которые возникают между любыми двумя атомами, входящими в фармпрепарат и биомолекулу, когда последние находятся на очень близком расстоянии, не превышающем 0,2 нм. Водородные связи имеют наиболее важное значение в действии фармакологических веществ и возникают только в том случае, если участвующий в ее образовании атом располагается на одной прямой со группой -ОН или -NH и на расстоянии не более 0,3 нм.Ионные связи (солеобразующие) возникают между ионами с разноименными зарядами, имея определенное значение для ассоциации лекарственного вещества с рецепторами тканей.
Важную роль в фармакологических и биохимических реакциях играет ион-дипольная связь, ориентирующая молекулы фармпрепарата относительно функционально активной группы фермента или рецептора. Для многих неионизированных молекул лекарственных веществ характерен дипольных момент. Некоторые атомы несут частичный (дробный), положительный или отрицательный заряд. Таким образом возникает полярность молекул. Диполь-дипольные связи принимают участие в фиксации лекарственного вещества на функциональной группе рецепторного поля.
Наиболее прочной связью является ковалентная, образующаяся между двумя атомами за счет общей пары электронов. Именно к резонансу электронов между атомами относят энергию ковалентной связи, которая возникает при взаимодействии мышьяка с SH–содержащими ферментами, фосфорорганических веществ с холинэстеразой, висмута и других тяжелых металлов с белками.
Первоначальным этапом реакции между лекарством и тканями организма является адсорбция, в основе которой лежит образование ван-дер-ваальсовых, водородных, ионных, дипольных связей. По-видимому, фармпрепарат притягивается рецептором, затем происходит ориентация его молекулы и, наконец, фиксация на рецепторном поле. Таким образом, специфический, характерный ответ клетки органа либо организма в целом развивается вслед за адсорбцией соединения на рецепторе. Если за адсорбцией происходит образование ковалентных связей, то имеет место очень прочная фиксация вещества на рецепторе, удалить которое физиологическим раствором нельзя.
📺 Видео
Фармакокинетика. Часть 1. Абсорбция. Распределение. 09.02.21Скачать

Клиническая фармакология №2 "Фармакокинетика лекарственных препаратов"Скачать

Статистика # 6. Показатели концентрации и дифференциацииСкачать

АГРЕГАТОМЕТРИЯ – Агрегация тромбоцитов/Агрегатограмма расшифровкаСкачать

Основы фармакокинетики. Олег ТалибовСкачать

Аскаридоз лошадейСкачать

Принципы назначения и интерпретации лабораторных исследованийСкачать

Фармакокинетическое взаимодействие ЛС на этапе всасыванияСкачать

1.2. Основы концепции биоэквивалентностиСкачать

Антибактериальные ЛП часть 2. 20.05.19Скачать

ВелоэргометрияСкачать

01 ФармакокинетикаСкачать

Клиническая фармакологии кардиологических препаратов . 17.09.20Скачать

Статистика. Урок 6. Показатели концентрации и дифференциацииСкачать

Поверхностно-активные веществаСкачать

НПВП - особенности применения при боли в суставахСкачать

3.1. История биоэквивалентности: с 1970-х годов по настоящее времяСкачать
